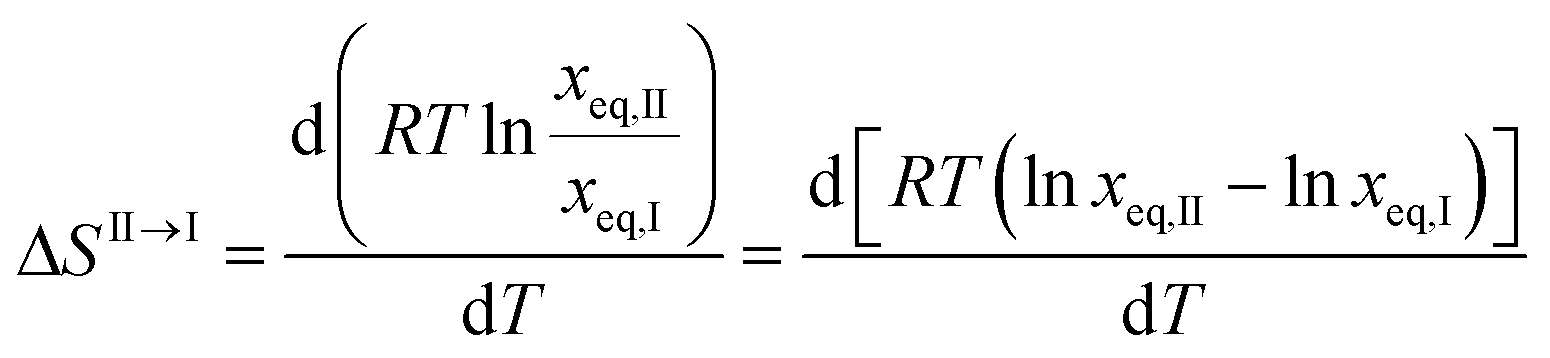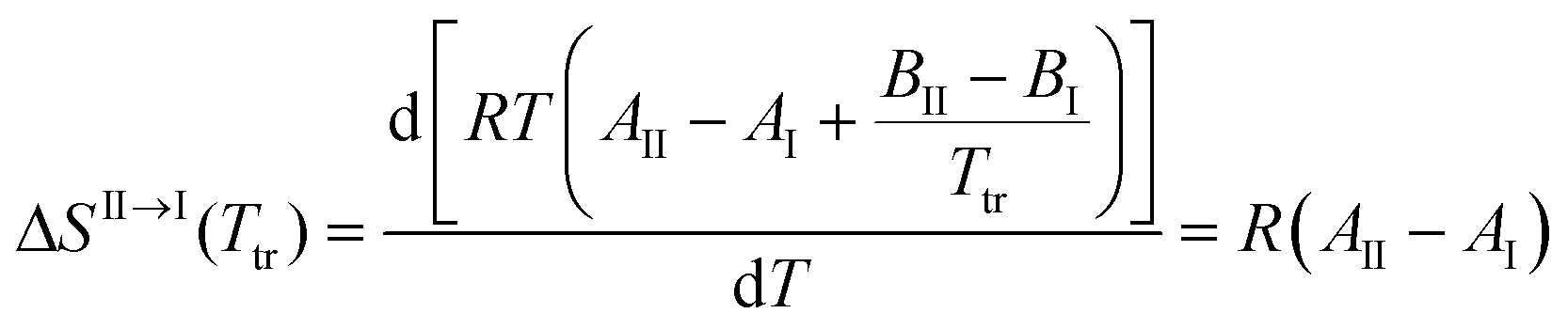DOI:
10.1039/C5CE00147A
(Paper)
CrystEngComm, 2015,
17, 3985-3997
Investigation into solid and solution properties of quinizarin†
Received
22nd January 2015
, Accepted 21st April 2015
First published on 21st April 2015
Abstract
Polymorphism, crystal shape and solubility of 1,4-dihydroxyanthraquinone (quinizarin) have been investigated in acetic acid, acetone, acetonitrile, n-butanol and toluene. The solubility of FI and FII from 20 °C to 45 °C has been determined by a gravimetric method. By slow evaporation, pure FI was obtained from n-butanol and toluene, pure FII was obtained from acetone, while either a mixture of the two forms or pure FI was obtained from acetic acid and acetonitrile. Slurry conversion experiments have established an enantiotropic relationship between the two polymorphs and that the commercially available FI is actually a metastable polymorph of quinizarin under ambient conditions. However, in the absence of FII, FI is kinetically stable for many days over the temperature range and in the solvents investigated. FI and FII have been characterized by infrared spectroscopy (IR), thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), scanning electron microscopy (SEM), transmission and ordinary powder X-ray diffraction (PXRD) at different temperatures. The crystal structure of FII has been determined by single-crystal XRD. DSC and high-temperature PXRD have shown that both FI and FII will transform into a not previously reported high-temperature form (FIII) around 185 °C before this form melts at 200–202 °C. By indexing FIII PXRD data, a triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) cell was assigned to FIII. The solubility of quinizarin FI and FII in the pure organic solvents used in the present work is below 2.5% by weight and decreases in the order: toluene, acetone, acetic acid, acetonitrile and n-butanol. The crystal shapes obtained in different solvents range from thin rods to flat plates or very flat leaves, with no clear principal difference observed between FI and FII.
cell was assigned to FIII. The solubility of quinizarin FI and FII in the pure organic solvents used in the present work is below 2.5% by weight and decreases in the order: toluene, acetone, acetic acid, acetonitrile and n-butanol. The crystal shapes obtained in different solvents range from thin rods to flat plates or very flat leaves, with no clear principal difference observed between FI and FII.
Introduction
Crystallization is a key process in the manufacturing of many organic compounds and of most pharmaceutical compounds.1 The solid–liquid solubility of the compound in different solvents is important information in the planning and design of the process. In addition, the solubility relationships carry information about how the solute molecule interacts with the solvent in solution and is accordingly of value in the interpretation of the influence of the solvent on nucleation and crystal growth.2 The crystal shape is of significant importance for downstream processing and end-use.3 Polymorphism has been an important topic for 20 years, especially in the pharmaceutical industry, where there is a requirement to identify all possible solid forms of a drug and where polymorphs can be protected by IP rights.4
Anthraquinones are the most important quinone derivatives of anthracene5 which are known to be present in many plant families such as Leguminosae, Liliaceae, Polygonaceae, Rubiaceae, and Rhamnaceae.6 Anthraquinone derivatives have been widely used as dyes for many years since they can provide a wide range of colours covering the entire visible spectrum.7 Anthraquinone derivatives exert a wide range of biological activities including laxative,8 diuretic,9 antioxidant,10 antifungal,11 antimicrobial and antiviral activities.12 They have also been used as anticancer agents, e.g. Ametantrone [64862-96-0] and Mitoxantrone (also known as Novantrone) [65271-80-9], to treat breast cancer and acute leukemia. Recently, more and more derivatives have been found to possess abilities as listed above, especially in the search of novel or second-generation anti-cancer drugs.13,14
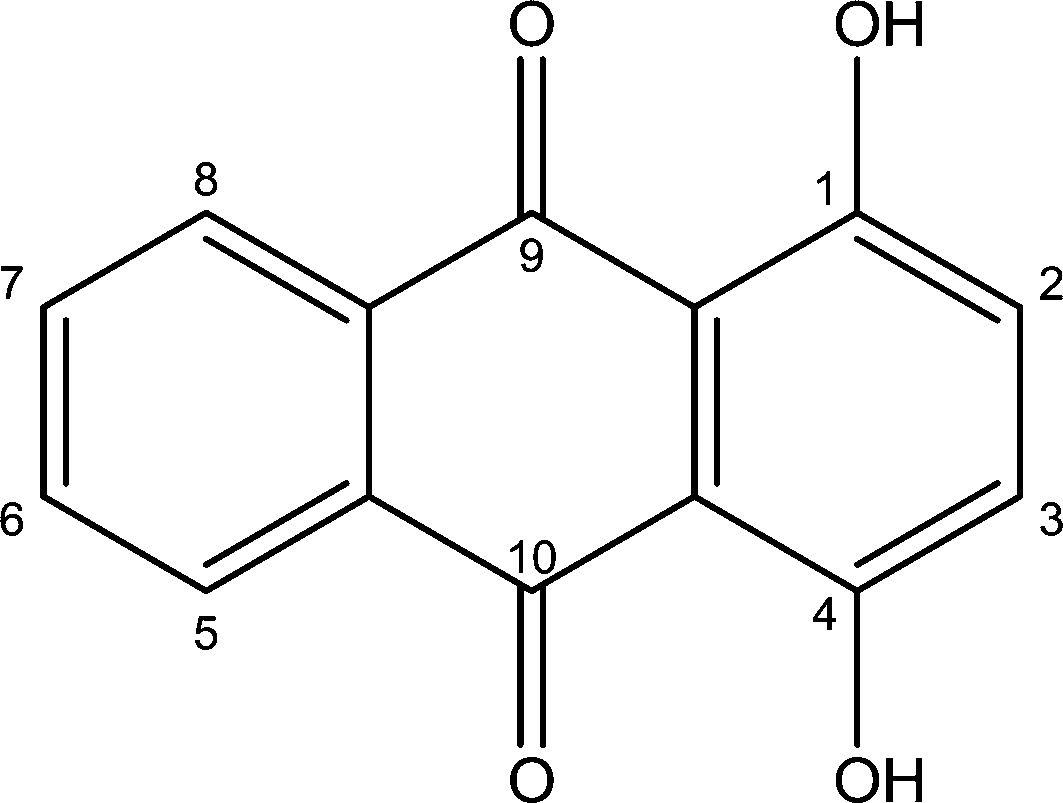
Among the anthraquinones, dihydroxyanthraquinones constitute the most important group and are largely used as dyes and in the manufacture of dye intermediates.15,16 They also form the building blocks of many medicinal drugs.17 1,4-Dihydroxyanthraquinone (quinizarin) (Fig. 1) is used as a fungicide and pesticide chemical18 and has shown the ability to inhibit tumour cell growth.19
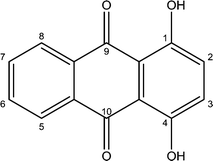 |
| | Fig. 1 Molecular structure of quinizarin. | |
Two polymorphs of quinizarin (FI and FII) are known, and FI has been reported as the stable form in the literature.20 Unit cells of both FI and FII have been reported.21 The full crystal structure of FI has been solved22 (CSD refcode DHXANT10) and it crystallizes in the monoclinic system, space group number 14 with one molecule per asymmetric unit. To date, to the best of our knowledge, no single crystal data for FII have been reported, and there are no published solubility data for quinizarin in organic solvents.
In this study, the solubility of both polymorphs in several pure organic solvents at different temperatures was measured using a gravimetric method. In crystallization experiments, different crystal shapes and polymorphic forms were investigated. In slurry conversion experiments, we show that there is, in fact, an enantiotropic relationship between FI and FII. In order to gain an understanding of the relationship between crystal structure and crystal growth, the crystal structure of FI was re-determined and the structure of FII has been solved, and in addition, crystals of each polymorph have been face indexed.
Experimental work
Materials
1,4-Dihydroxyanthraquinone (CAS reg. no. 81-64-1, purity 99.3%, verified to be FI by transmission PXRD) was purchased from Fluka and used without further purification. FII used in this work was obtained by slurrying excess FI in acetone at 10 °C. Organic solvents were purchased from Sigma-Aldrich and used as received: acetic acid (Puriss grade, ≥99.8%), acetone (Chromasolv, ≥99.8%), acetonitrile (Chromasolv plus, ≥99.9%), n-butanol (Chromasolv plus, ≥99.7%) and toluene (Chromasolv, 99.9%).
Experimental setup
The setup consisted of a thermostatic water bath (Grant S26 stainless steel water bath; 26 L, 505 × 300 × 200 mm; equipped with a Grant C2G cooling unit and a Grant GR150 control unit; at 310 K, stability ±0.005 K and uniformity ±0.02 K) with a serial submersible 60 points magnetic stirrer plate (2Mag) placed on the base and a submersible water pump (1400 L h−1) to enhance circulation in the bath. The set temperature was validated by a digital thermometer (D750 PT, Dostmann electronic, Wertheim, Germany, uncertainty of ±0.01 K).
Thermodynamic and kinetic stability
Mechanical mixtures of solid FI and FII were slurried in the five organic solvents and at different temperatures from 30 °C to 80 °C in 10 °C increments, and the sampled solids were analysed with transmission PXRD. Saturated solutions with excess FI in different solvents were prepared in 30 mL glass vials with PTFE-coated magnetic stirrers, at 25 °C, 35 °C and 50 °C, and the solids were sampled and analysed with transmission PXRD after five days and ten days.
Solubility of FI and FII
Solutions with excess pure FI or FII were prepared in 30 mL glass vials with PTFE-coated magnetic stirrers. The vials were placed on the magnetic stirrer plate with agitation of 600 rpm in the water bath for at least 18 hours at each temperature to allow equilibrium to be reached. Stirring was then turned off and excess solids were allowed to settle for two hours. Clear solution was sampled using syringes (5 mL) and filtered through syringe filters (PTFE/Nylon, 25 mm, pore size 0.2 μm, VWR) into pre-weighed glass vials. Syringes and filters were pre-heated to 5 °C above the saturation temperature to prevent nucleation inside the syringes during sampling. The weight of each solution was recorded immediately. The samples were completely dried in a ventilated laboratory hood at room temperature, and the weight of each sample was recorded repeatedly throughout the drying process in order to ensure complete dryness. Finally, the samples were dried in an oven at 40 °C for six hours to ensure all solvent had evaporated. At each temperature, the solids in equilibrium with the solution were sampled, dried and analysed by ex situ PXRD in order to ensure that no transformation of the original solid structure had occurred.
Crystal shape
Slow evaporation crystallisation experiments were performed in 5 mL scale at room temperature in a ventilated laboratory hood. The solvent was allowed to evaporate through small holes in the aluminium foil cover. Cooling crystallisation experiments were conducted in 30 mL vials filled with approximately 20 mL solution saturated at 40 °C and agitated at 600 rpm using magnetic stirrers. Solutions were cooled instantaneously from 45 °C to either 35 °C or 5 °C. In some experiments, agitation was suspended immediately upon visible nucleation. The crystals were sampled twice by filtration, initially after nucleation occurred and subsequently after five days under the same conditions. Crystals were analysed using PXRD and SEM.
Solid-state characterization
Solid samples of quinizarin have been identified and characterized using attenuated total reflection infrared spectroscopy (ATR-IR, Perkin Elmer Spectrum 100 IR spectrometer with a Universal ATR sampling accessory), transmission powder X-ray diffraction (PXRD, PANalytical Empyrean) and variable-temperature PXRD (PANalytical X'Pert MPD Pro with a hot stage accessory). Crystal shape analysis has been carried out using scanning electron microscopy (SEM, JEOL CarryScope scanning electron microscope JCM-5700). The thermal behaviour has been investigated using differential scanning calorimetry (DSC, PerkinElmer Pyris 1) and thermogravimetric analysis (simultaneous DSC-TGA, SDT, Q600, TA Instruments).
Single-crystal X-ray diffraction
An Oxford Diffraction Xcalibur system was used to collect X-ray diffraction data. The crystal structures of FI and FII were solved by direct methods (Shelxs) and refined by full matrix least-squares using Shelxl-2014 within the Oscail package.23,24 The crystal structure of FI was refined close to the orthogonal P21/n setting with a β angle of 95.999(8)°, while the structure of FI in the CSD with refcode DHXANT10 was refined in P21/a with an obtuse β angle of 125.46°. It is preferable to use the more orthogonal cell.25
Results and discussion
Solvent selection and polymorph screening
A common approach for investigating the polymorphic behaviour of a compound is to perform solution crystallizations using a range of solvents chosen to have diverse properties in order to maximise the chances of isolating different solid forms. Here, a set of five solvents with relatively higher solubility (predicted using Hansen solubility parameters) was selected, having a range of polarities and functional groups for solution crystallizations. Crystallizations including slow evaporation, cooling with different cooling rates and slurry conversion were performed. In addition, DSC and thermogravimetric analysis were carried out to determine the thermal behaviour of quinizarin. Three forms of quinizarin were found, but only FI and FII were obtained under ambient conditions. FI and FII are enantiotropically related to each other, and both appear as dark orangey-red plates or needles.
Crystal structures of FI and FII
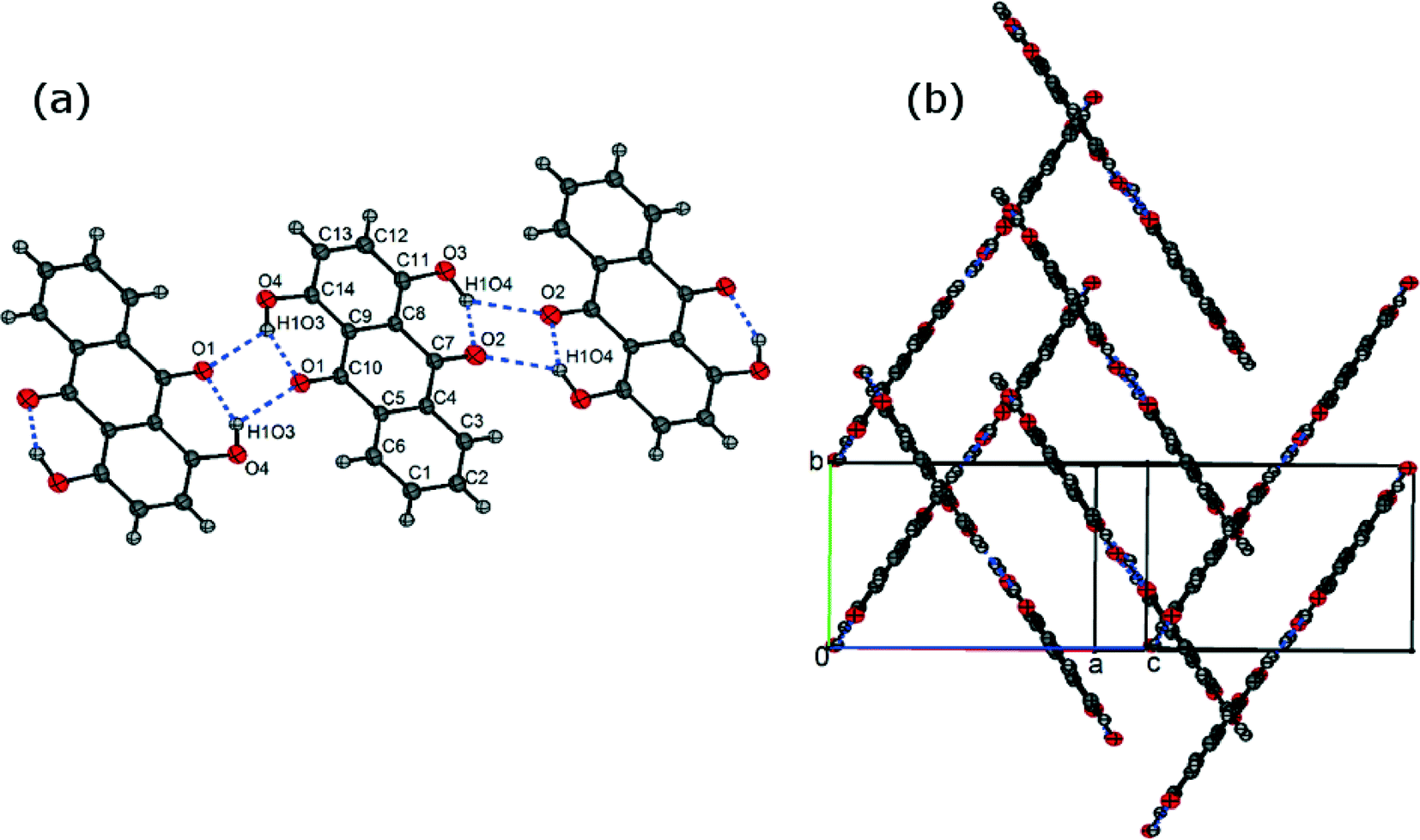
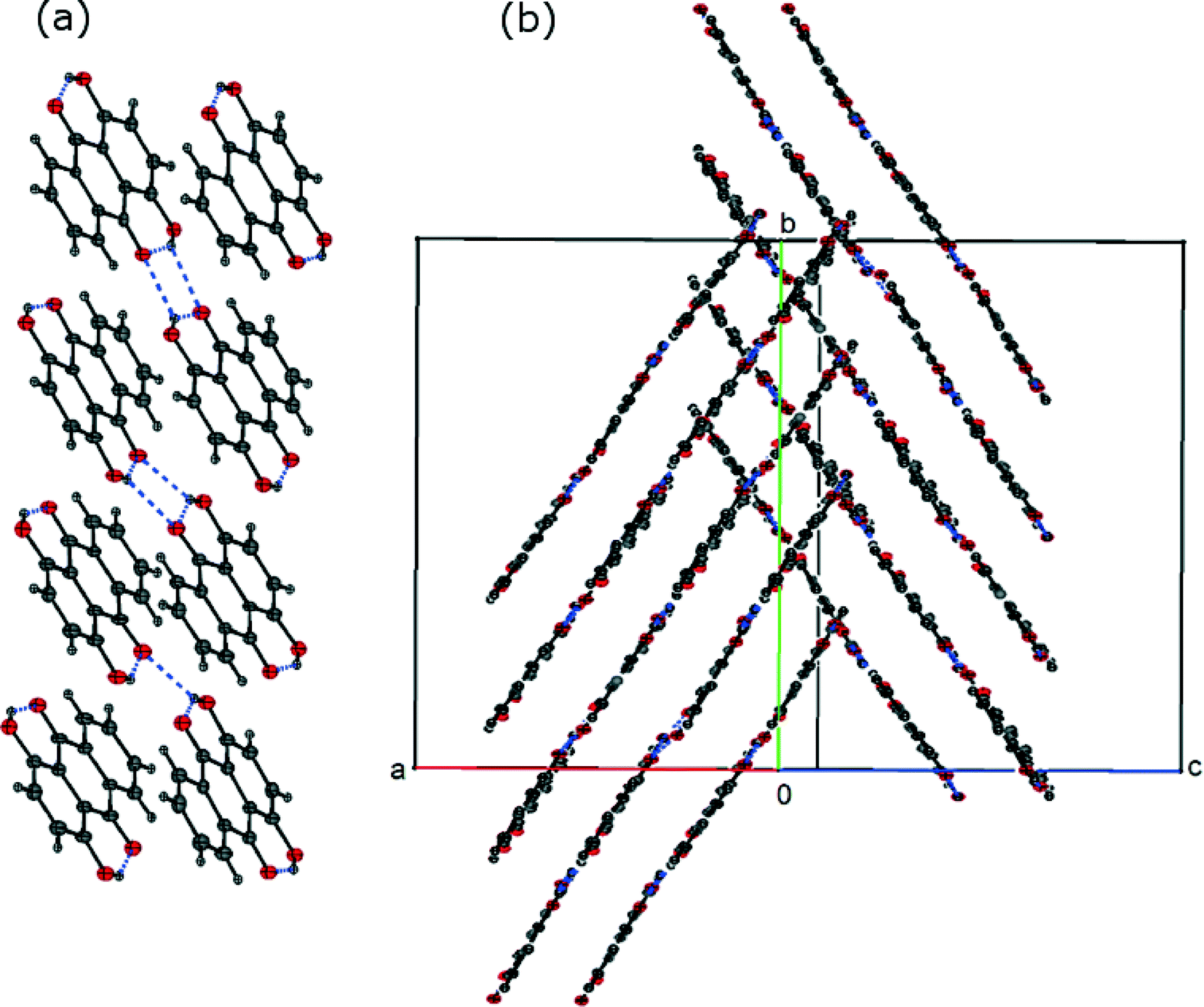
The crystal structure of FI was determined by single-crystal XRD, using a needle-shaped crystal grown from toluene solution. In the FI structure, the molecules form intra- and intermolecular hydrogen bonds leading to the formation of ribbons (Fig. 2(a)). These ribbons are arranged in slipped π-stacks which run along the b axis (Fig. 2(b)).
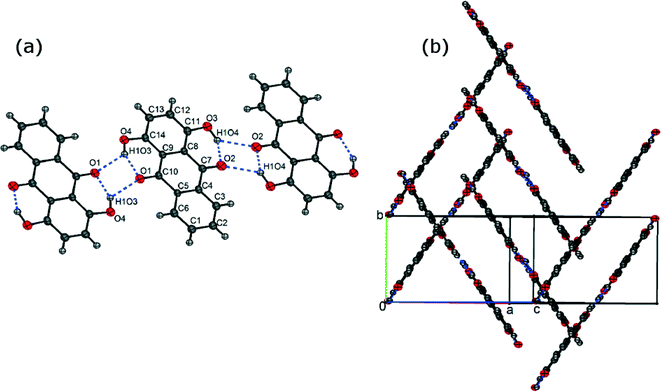 |
| | Fig. 2 (a) Asymmetric unit of the FI structure (labelled atoms) and its hydrogen bond ribbon motif and (b) π-stacking in the FI structure. | |
The crystal structure of FII was determined using a needle-shaped crystal grown from acetic acid solution. The asymmetric unit containing 8 molecules is shown in Fig. 3(a). As in the FI structure, the molecules form intra- and intermolecular hydrogen bonds leading to the formation of ribbons which are stacked along the b axis (Fig. 3(b)).
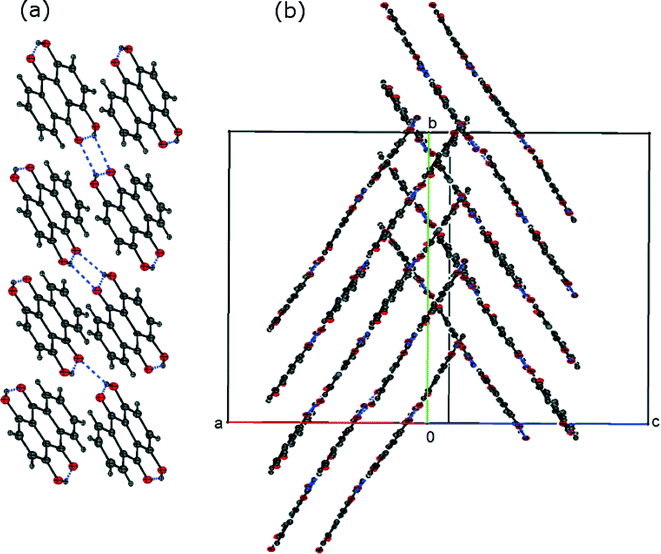 |
| | Fig. 3 (a) Asymmetric unit of the FII structure and (b) π-stacking in the FII structure. | |
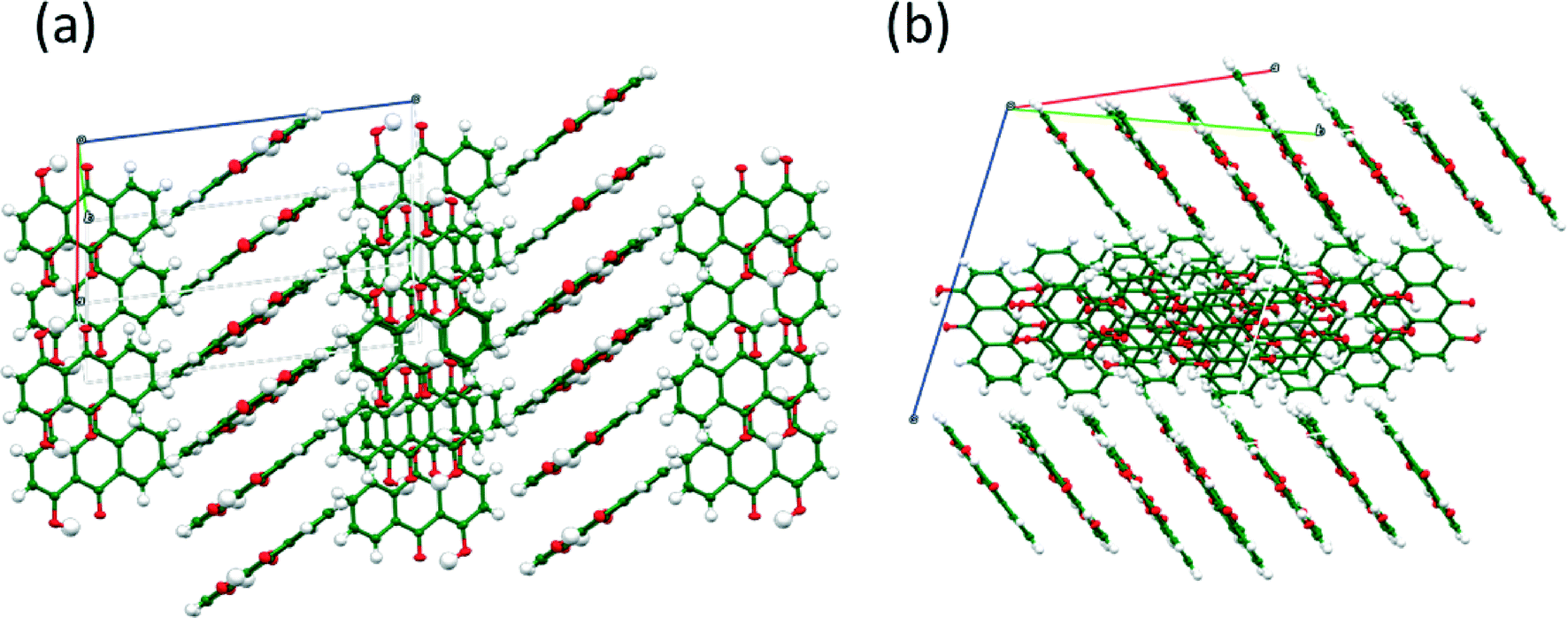
The structures of FI and FII contain essentially the same bonding motifs, and the difference between the two crystal structures is in the packing of the 1-D chains of quinizarin molecules. FI forms a herringbone packing with alternating stacks (Fig. 4(a)). In contrast, FII forms a stack of molecules in alternating layers (Fig. 4(b)). The transformation from FI to FII involves a reduction in the symmetry of FI in which eight symmetry-related molecules become independent to accommodate tiny changes in conformation which allow the FII structure to pack better and give a small increase in density. Crystal structure data and hydrogen bond distances of both structures are provided in the ESI.†
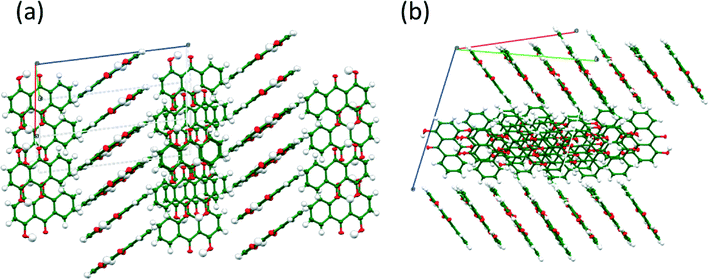 |
| | Fig. 4 (a) View of the herringbone packing of FI, viewed along the b-axis and (b) view of the packing of layers in FII. | |
Characterisation of polymorphic forms
Mid-infrared spectra of FI and FII are quite similar, and the unique regions of these two forms are below 1700 cm−1 and only slight differences can be spotted at 671, 772–784 and 1176 cm−1. The high similarity in IR spectra was expected considering the small difference in terms of hydrogen bonding between the two forms. The IR spectrum of the commercial FI is identical to the spectra reported in the literature26,27 and the spectrum has been discussed by Nasrallah et al.28 and Xuan et al.29 IR spectra of FI and FII are provided in the ESI.†
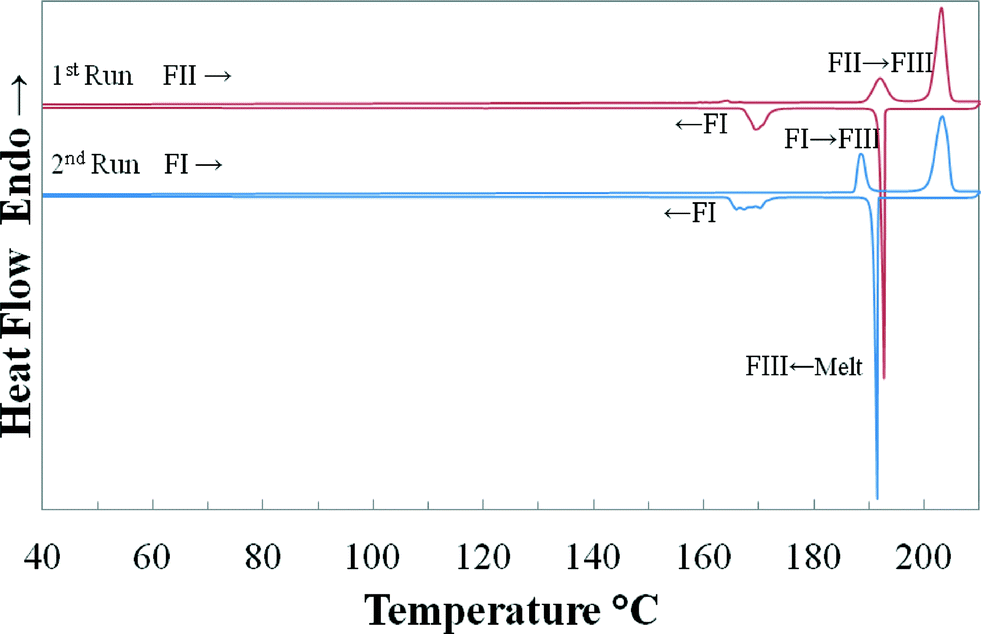
Fig. 5 shows DSC thermograms from two consecutive heating–cooling cycles with a ramp rate of 10 K·min−1, starting with FII and FI, respectively. The cycles have been repeated several times in order to isolate and identify the polymorphic form at different transformation stages using PXRD. It can be seen that both FI and FII transformed with an endothermic peak into a previously unreported high-temperature stable polymorph (FIII) at 185.8 °C and 189.3 °C, respectively. FIII started to melt at approximately 200 °C. Upon cooling, the melt recrystallized around 190 °C followed by a further transformation into FI below 175 °C. Attempts made to isolate this high temperature form using different cooling rates and quenching the DSC pans containing the melt or FIII in liquid nitrogen have been unsuccessful, resulting in FI. The determined enthalpy, extrapolated onset and peak temperature of the observed thermal events, averaged over 6 scans, are given in Table 1.
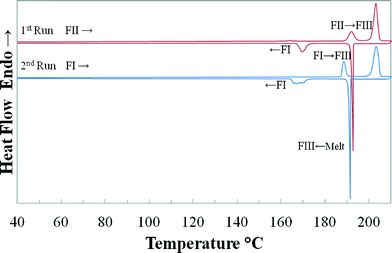 |
| | Fig. 5 DSC heating–cooling cycles (10 K min−1) using quinizarin FII as starting material, indicating the transformations into high-temperature FIII and the melting of FIII. | |
Table 1 DSC data of quinizarin reported with 95% confidence intervals
|
|
FI to FIII transformation |
FII to FIII transformation |
Melting of FIII |
|
T (onset)/°C |
185.79 ± 0.57 |
189.32 ± 0.30 |
200.00 ± 0.67 |
|
T (peak)/°C |
187.62 ± 0.29 |
191.64 ± 0.35 |
202.97 ± 0.19 |
| ΔH/kJ·mol−1 |
6.65 ± 0.058 |
6.81 ± 0.074 |
19.41 ± 0.14 |
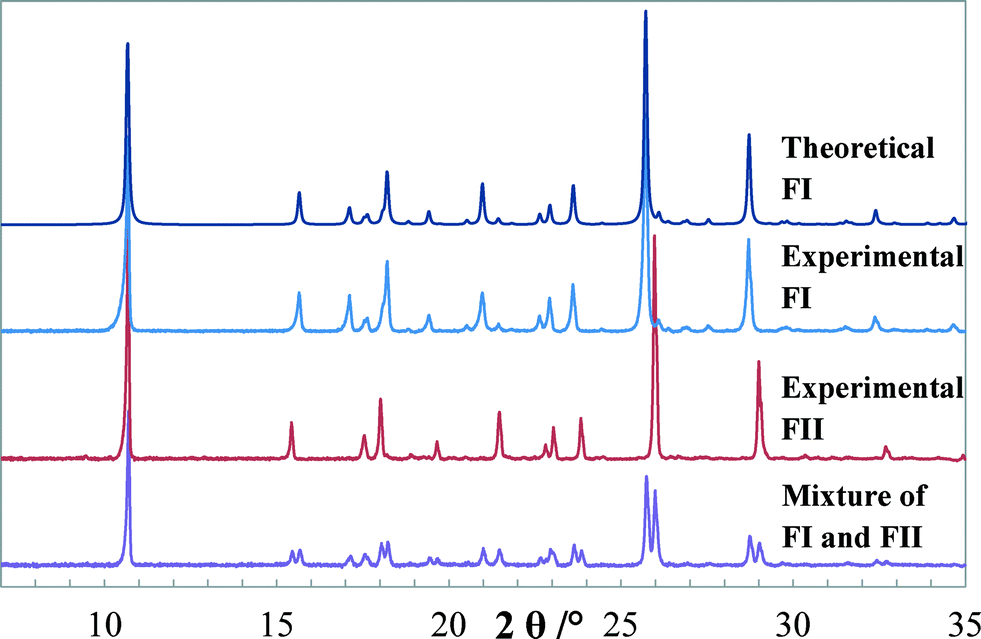
Transmission PXRD patterns of quinizarin, as shown in Fig. 6, demonstrate the commercial quinizarin to be pure FI, with an XRD pattern identical to the theoretical pattern of FI obtained from the structure DHXANT10. Prominent peak positions (2θ values) for FI are at 15.65, 17.12, 18.21, 20.97, 23.60, 25.70 and 28.71°, and for FII, they are at 6.0, 15.43, 18.02, 21.47, 23.83, 25.97 and 29.00°.
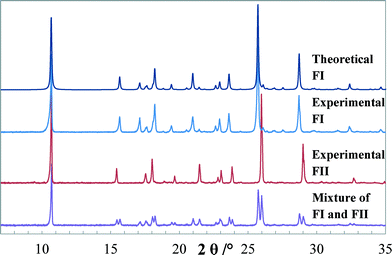 |
| | Fig. 6 Theoretical PXRD pattern of FI (DHXANT10) along with experimental PXRD patterns of FI, FII and a mechanical mixture of FI and FII. | |
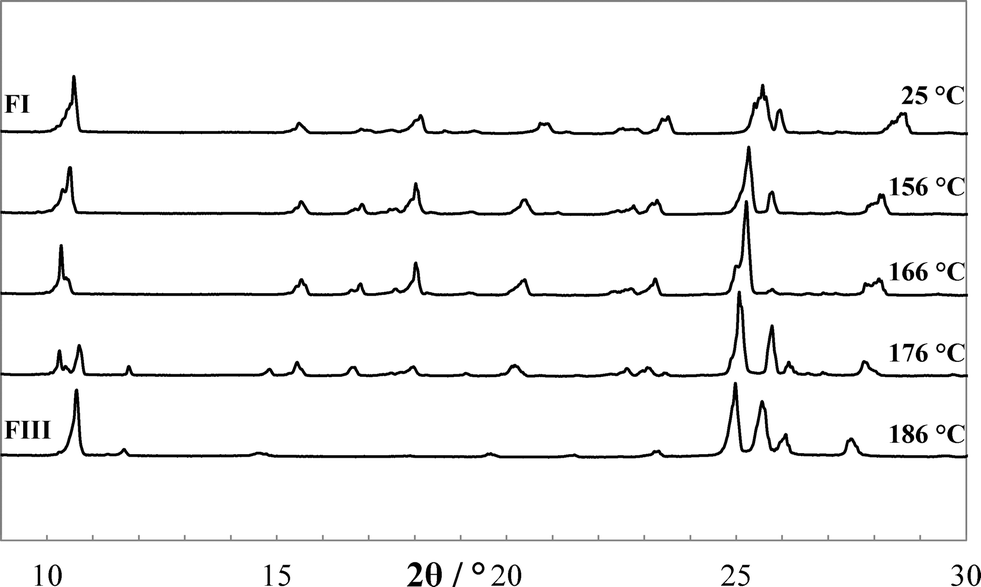
The endothermic transformation of FI into FIII was confirmed to be solid–solid phase transformation by applying different heating rates (0.2, 2, 10 and 50 K min−1) using DSC. This transformation was further validated by HT-PXRD, equipped with a hot stage accessory that was calibrated with indium. Visual comparison of the HT-PXRD data (Fig. 7) indicates that a new phase appeared at about 176 °C, with complete conversion by 186 °C. Reduction of diffraction angles can be observed as the temperature increases, due to the temperature dependence of lattice constants.
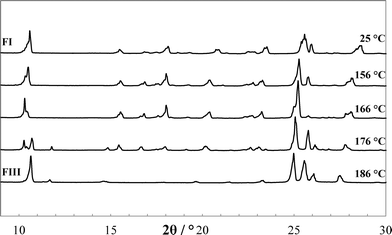 |
| | Fig. 7 HT-PXRD showing the high-temperature transformation of FI into FIII. | |
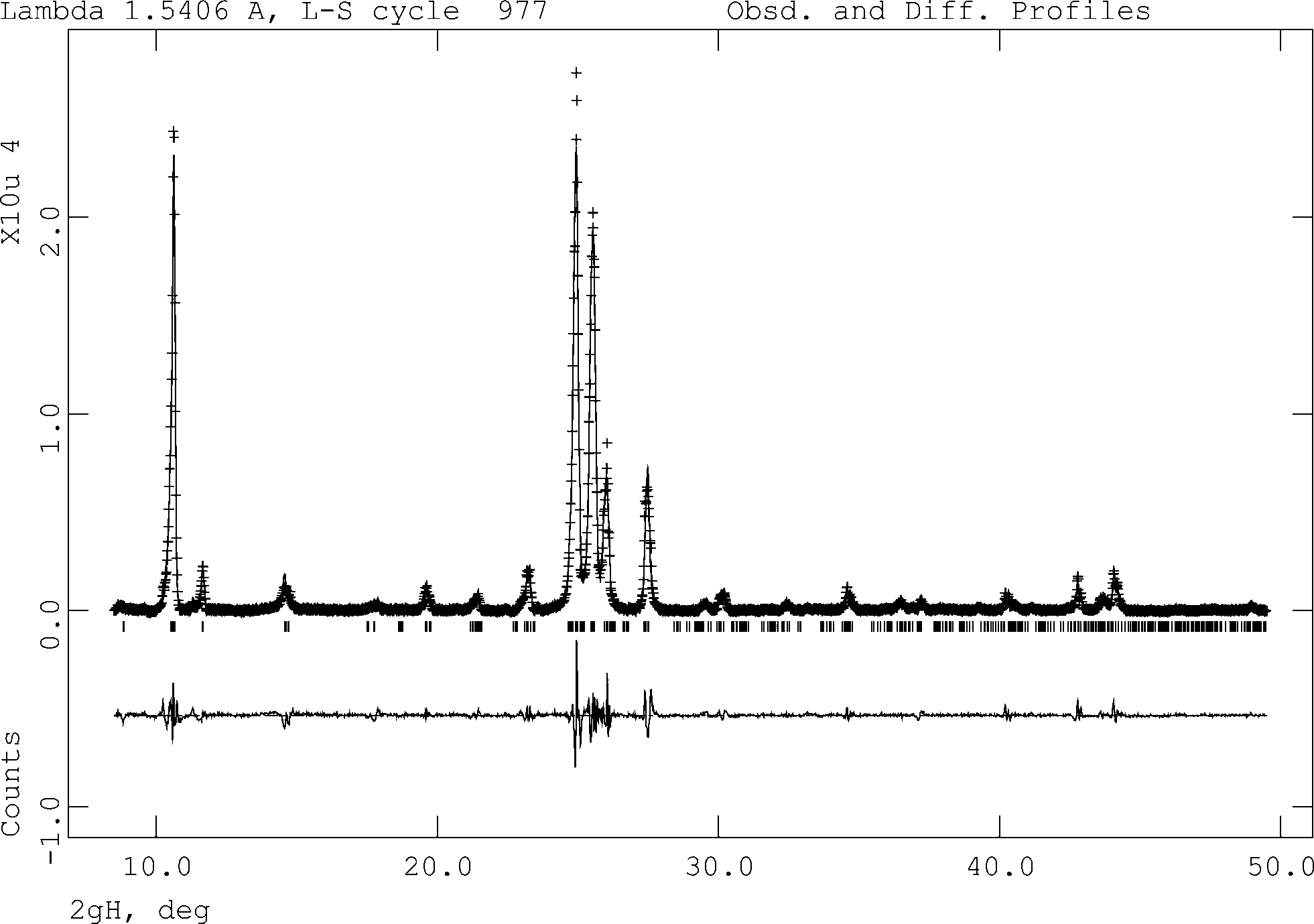
Applying Le Bail analysis using the program GSAS30 on the FIII PXRD data at 180 °C, it was found that none of the known unit cells (FI, FII from CSD and our determined structures) give a satisfactory fit to the experimental data. Therefore the obtained peak positions were used as the basis of indexing attempt using the range of indexing programs available in the CMPR program.31 Only the ITO indexing program32 located a suitable cell (a = 4.2846 Å, b = 10.1957 Å, c = 15.3383 Å, α = 88.6360°, β = 83.2210°, γ = 78.3660°, V = 651.69 Å3). While this displays a figure of merit lower than those normally considered correct (M20 = 5.4), it does match all the peaks in the pattern, and the cell parameters and volume are consistent with a molecule of this size (2 molecules in the unit cell assuming 18 Å3 for each non-H atom). Therefore a Le Bail analysis was attempted in P for this cell to confirm the match of the cell to experimental data. This was successfully carried out (Rwp = 7.75%, Rp = 5.60%, χ2 = 8.727, Fig. 8). A comparison of the three structures is shown in Table 2. The metric values of the unit cell are similar to the FII cell reported in the CSD (a = 14.80 Å, b = 9.49 Å, c = 3.77 Å, α = 90°, β = 93.0°, γ = 90°); however, this cell cannot be refined to fit the data as well as the indexed triclinic cell and so we believe the triclinic cell to be the correct high-temperature cell. This phase, reported as FII in the previous work by Borgen in 1966 (ref. 21), was different from our experimental FII and was obtained from slow cooling of the melt in which some crystals reverted to FI at room temperature. Thus this cell may refer to another high-temperature polymorph of quinizarin.
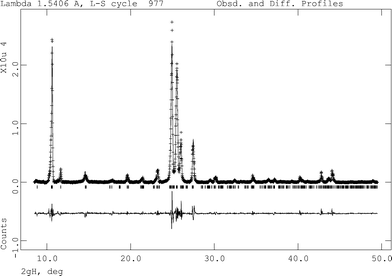 |
| | Fig. 8 Final Le Bail fit to PXRD data of FIII at 180 °C. | |
Table 2 Comparison of unit cell data for FI, FII and FIII data predicted by Le Bail analysis
|
|
FI |
FII |
Predicted FIII |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P21/n |
Cc
|
P |
| Unit cell dimensions |
a = 10.2390(8) Å |
a = 20.0099(12) Å |
a = 4.2846 Å |
|
b = 6.0429(4) Å |
b = 24.6219(9) Å |
b = 10.1957 Å |
|
c = 16.454(2) Å |
c = 18.3201(11) Å |
c = 15.3383 Å |
|
α = 90° |
α = 90° |
α = 88.6360° |
|
β = 95.999(8)°. |
β = 116.274(8)° |
β = 83.221° |
|
γ = 90° |
γ = 90° |
γ = 78.366° |
| Unit cell volume |
1012.51(17) Å3 |
8093.4(9) Å3 |
651.69 Å3 |
|
Z
|
4 |
32 |
2 |
|
Z′ |
1 |
8 |
1 |
| Density (calculated) |
1.576 Mg m−3 |
1.577 Mg m−3 |
1.22 Mg m−3 |
Influence of solvent on polymorph and crystal shape
Quinizarin crystals obtained from the five organic solvents by slow evaporation at room temperature exhibit differences in shape and polymorphic form. Recrystallization by slow evaporation at room temperature from acetone solution consistently produced pure FII, while pure FI was always obtained from n-butanol and toluene solutions. In samples obtained from acetic acid and acetonitrile solution, a mechanical mixture of FI and FII was often found. In cooling crystallizations from 40 °C to 35 °C, FI was always obtained from all different solvents. However, with a higher cooling temperature range from 40 °C to 5 °C in acetone, either pure FII or a mechanical mixture of FI and FII was obtained. Thermogravimetric analysis of the solid samples obtained from the five solvents all show a one-step decomposition process between 200 and 300 °C, suggesting that no solvates were formed.
Some of the crystal habits observed by SEM are shown in Fig. 9–11. By slow evaporation, thin plates were obtained from acetic acid and acetonitrile solutions, rods from acetone and toluene solutions, while a leaf-shaped habit was obtained from n-butanol (Fig. 9). In cooling crystallization (ΔT = 5 °C), quinizarin mainly crystallizes as plates or prisms (Fig. 10) but as needles in toluene solution when no agitation was used (Fig. 10(f)). Moreover, with a higher driving force (ΔT = 35 °C) in acetone (Fig. 11), quinizarin crystallizes as very thin plates and the morphology is undistinguishable between the two forms. From observation, quinizarin generally crystallizes as plates and needles and often seen mixed together without any tendency of the solvent effects. However, we can see that the needle growth is favoured without agitation and a high driving force will yield very thin plate crystals.
 |
| | Fig. 9 Crystals recrystallized by slow evaporation in (a) acetic acid (mix of FI and FII), (b) acetone (FII), (c) acetonitrile (mix of FI and FII), (d) n-butanol (FI) and (e) toluene (FI). | |
 |
| | Fig. 10 FI recrystallized by cooling (ΔT = 5 °C) in (a) acetic acid, (b) acetone, (c) acetonitrile, (d) n-butanol, (e) toluene with agitation and (f) toluene without agitation. | |
 |
| | Fig. 11 (a) Mixture of FI and FII and (b) pure FII obtained by fast cooling (ΔT = 35°C) in acetone. | |
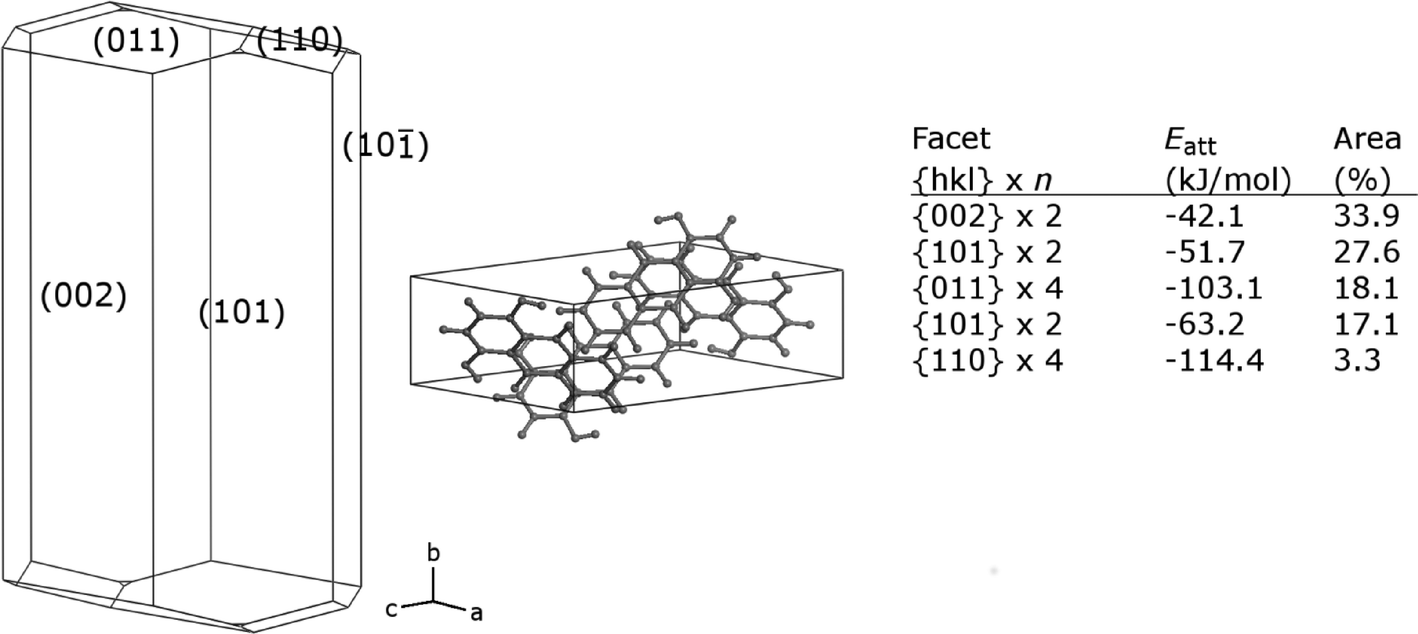
A prediction of the crystal morphology of FI in vacuo was obtained using the attachment energy method.33 These calculations assume that the growth rates of different faces are based only on the energy released in forming the lattice and that kinetic factors including solvent effects make no contribution. Calculations were carried out using the Morphology module of Materials Studio 7.0 (Accelrys), with the pcff force field34 and force field-specific point charges. Fig. 12 shows the resulting crystal shape, with Miller indices of visible faces. FI is predicted to favour a plate-like habit, elongated in the b-direction, dominated by the two {002} faces with the lowest attachment energy, followed by the two {101} faces. The predicted habit is more prismatic than observed experimentally, which emphasizes that kinetic factors can dominate crystal growth from solution.
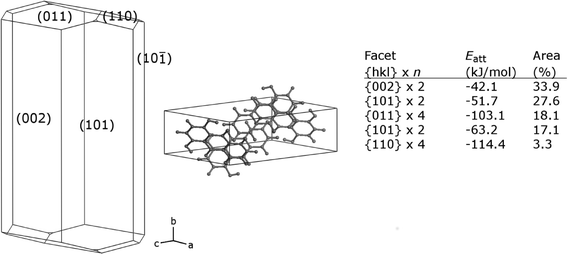 |
| | Fig. 12 Predicted vacuum crystal morphology of FI, with unit cell representation and tabulated data for dominant faces. | |
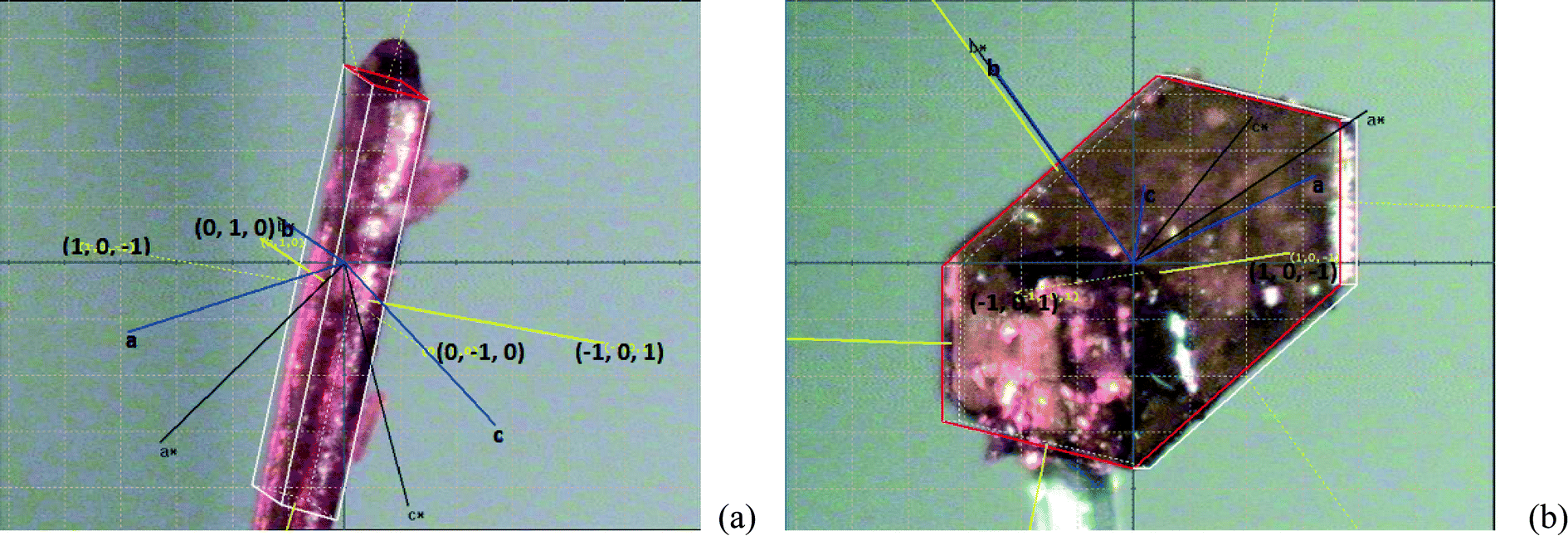
Using single-crystal XRD, the main visible faces of a needle-shaped crystal of FI grown from toluene solution were indexed (Fig. 13). The needle growth direction in the FI crystal structure is along the b axis, as predicted, coinciding with the direction of π-stacking of ribbons. The planes defined by the ribbons are 3.35 Å apart with the atoms of adjacent ribbons in van der Waals contact and this is typical of slipped π-stacked systems and similar to the packing in β-phthalocyanine and benzoic acid.35 The high growth rate in the needle direction of β-phthalocyanine has been associated with the low activation energy required to generate a new step in the stacking direction.36 The importance of the crystallization driving force in controlling needle growth in the gas phase for flat molecule stacked systems has also been demonstrated.37
 |
| | Fig. 13 Face indexed crystal of FI from toluene solution, showing needle growth along the b axis. | |
Notably, in the present case, when agitation was suspended upon nucleation needles were observed (Fig. 10(f)), whereas if agitation continued after nucleation the crystals became more blocky. The possibility of this being the result of crystal breakage due to agitation was ruled out based on visual observation. Without agitation, the effective supersaturation is expected to decrease locally and thus result in a reduced crystallization driving force close to the growing crystal surface, favouring needle growth.35 It might appear surprising that this system in solution mimics the behaviour of other flat molecule systems in the gas phase. However, toluene is not a hydrogen bonding solvent and would not be expected to exert a direct influence on the crystal growth.
Face-indexed crystals of FII grown from acetic acid without agitation are shown in Fig. 14. The crystals are initially needle-like (Fig. 14(a)) but with time gradually take on a more isotropic shape (Fig. 14(b)). The needles show extended growth in directions normal to the {101} faces. In the more prismatic crystals which develop after some time the {10−1} faces are dominant. It is clear that unlike FI growth in toluene, FII growth in acetic acid solution is not controlled by stacking effects. Here solvent hydrogen bonding to growing crystal faces seems to be more important.
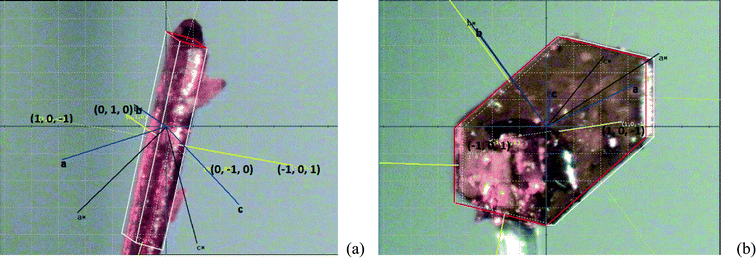 |
| | Fig. 14 Face indexed crystals of FII from acetic acid solution: (a) initially observed needle and (b) later developed, more prismatic crystal. | |
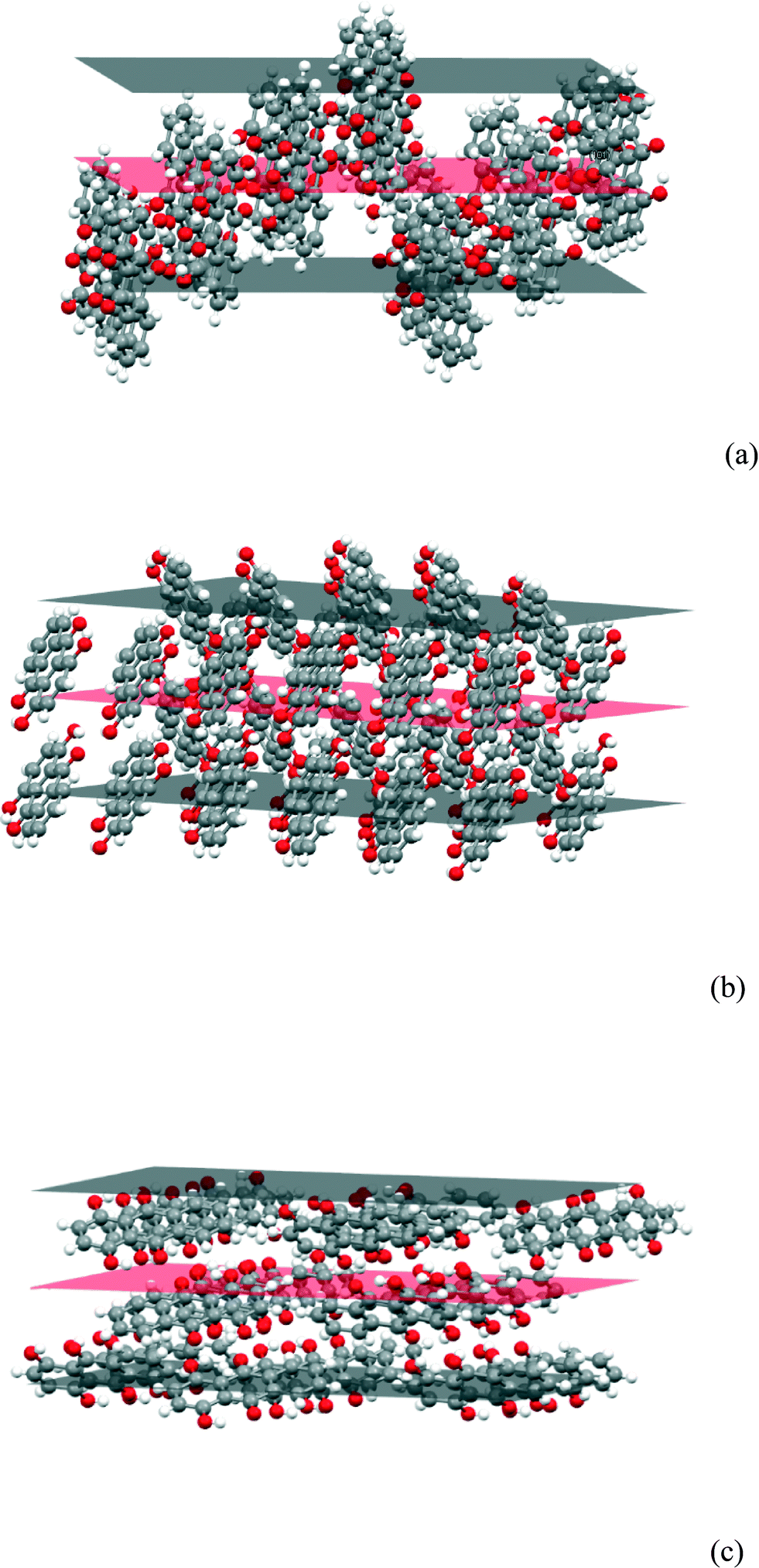
If the packing at the {101}, {010} and {10−1} faces are compared (Fig. 15), it is clear that the hydrogen bonding solvent will bind more strongly to the latter faces which have a higher concentration of exposed O atoms and OH groups. The solvent molecules would be expected to bind least strongly to the {101} faces as they are dominated by surface CH groups and it is reasonable that its growth is initially faster. It is also reasonable that the {10−1} faces are the ones which eventually become dominant as they provide the greatest surface concentration of O atoms and OH groups and thus are likely to give the lowest interfacial energy.
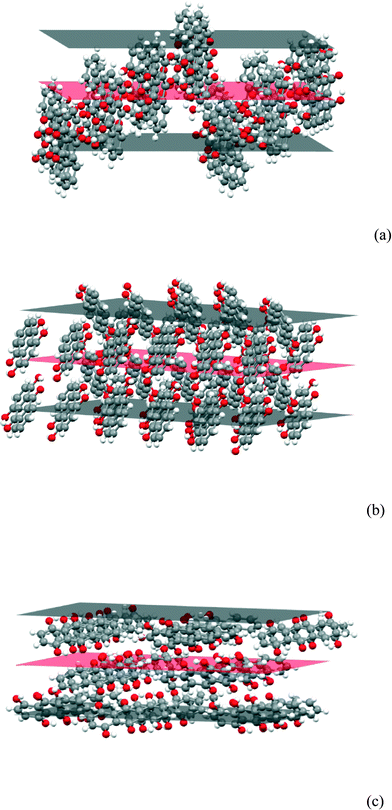 |
| | Fig. 15 The (a) (101), (b) (010) and (c) (10−1) faces of the FII structure. | |
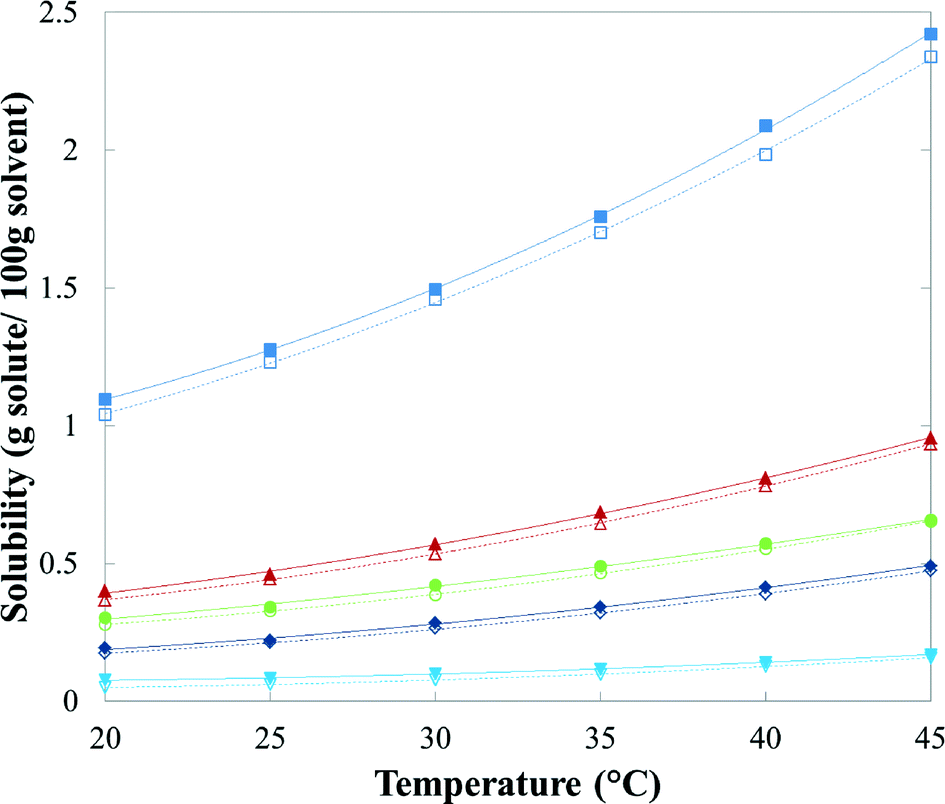
Solubility and thermodynamic stability relationship
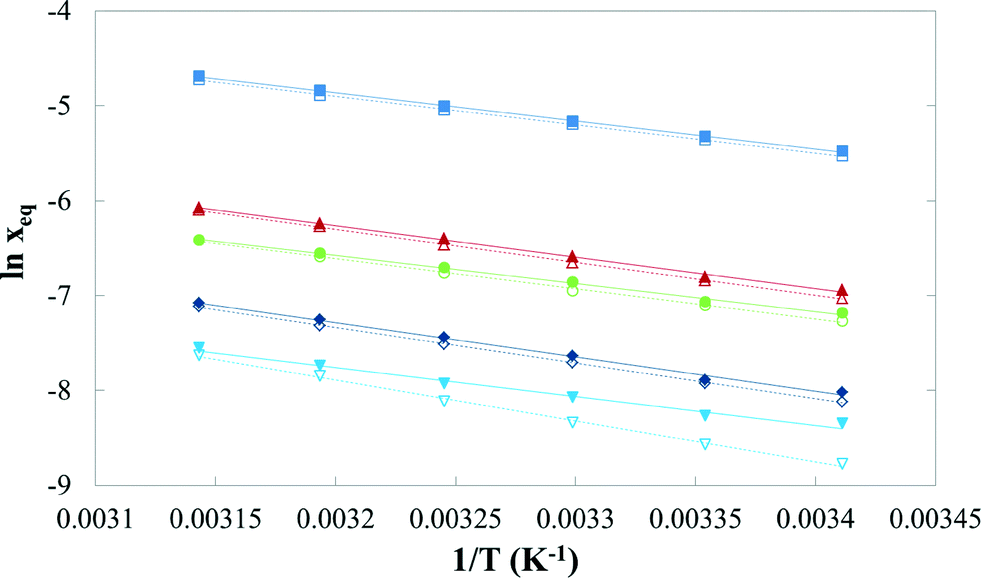
Table 3 presents mean values and standard deviations of triplicate measurements of the solubility of FI and FII in five pure solvents at temperatures between 20 °C and 45 °C. The standard deviations are relatively small (less than 2.24 × 10−2 g per 100 g of solvent in all cases) indicating good reproducibility. The data are represented graphically in Fig. 16, with linear axis scales, and as a van't Hoff plot in Fig. 17, in which the logarithm mole fraction solubility, ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) xeq, is plotted against the reciprocal of the absolute temperature.
xeq, is plotted against the reciprocal of the absolute temperature.
Table 3 Solubility of 1,4-dihydroxyanthraquinone (FI and FII) in five solvents from 20 °C to 45 °C
| Solubility of quinizarin FI (standard deviation over three samples) (g per 100 g of solvent) |
|
T/°C |
Acetic acid |
Acetone |
Acetonitrile |
n-Butanol |
Toluene |
| 20 |
0.3036 (0.0004) |
0.3986 (0.0045) |
0.1924 (0.0033) |
0.0768 (0.0046) |
1.0968 (0.0044) |
| 25 |
0.3417 (0.0007) |
0.4601 (0.0045) |
0.2198 (0.0005) |
0.0833 (0.0035) |
1.2769 (0.0143) |
| 30 |
0.4208 (0.0038) |
0.5713 (0.0031) |
0.2836 (0.0046) |
0.1012 (0.0032) |
1.4951 (0.0011) |
| 35 |
0.4912 (0.0044) |
0.6865 (0.0045) |
0.3433 (0.0063) |
0.1169 (0.0054) |
1.7583 (0.0049) |
| 40 |
0.5730 (0.0051) |
0.8101 (0.0086) |
0.4144 (0.0015) |
0.1416 (0.0033) |
2.0879 (0.0154) |
| 45 |
0.6573 (0.0123) |
0.9555 (0.0177) |
0.4915 (0.0060) |
0.1702 (0.0019) |
2.4204 (0.0224) |
| |
| Solubility of quinizarin FII (standard deviation over three samples) (g per 100 g of solvent) |
|
T/°C |
Acetic acid |
Acetone |
Acetonitrile |
n-Butanol |
Toluene |
| 20 |
0.2790 (0.0008) |
0.3677 (0.0007) |
0.1745 (0.0022) |
0.0503 (0.0005) |
1.0394 (0.0025) |
| 25 |
0.3287 (0.0007) |
0.4440 (0.0027) |
0.2122 (0.0004) |
0.0615 (0.0012) |
1.2296 (0.0119) |
| 30 |
0.3854 (0.0068) |
0.5350 (0.0003) |
0.2638 (0.0015) |
0.0776 (0.0005) |
1.4601 (0.0084) |
| 35 |
0.4649 (0.0027) |
0.6447 (0.0022) |
0.3188 (0.0010) |
0.0974 (0.0011) |
1.7004 (0.0095) |
| 40 |
0.5526 (0.0007) |
0.7826 (0.0042) |
0.3897 (0.0017) |
0.1270 (0.0013) |
1.9837 (0.0018) |
| 45 |
0.6540 (0.0011) |
0.9341 (0.0023) |
0.4752 (0.0008) |
0.1580 (0.0005) |
2.3399 (0.0115) |
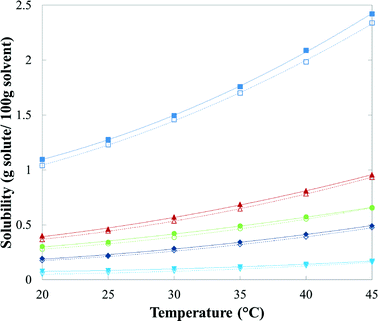 |
| | Fig. 16 Solubility of quinizarin FI (solid symbols) and FII (hollow symbols) in: ■, toluene; ▲, acetone; ●, acetic acid; ◆, acetonitrile; and ▼, n-butanol. | |
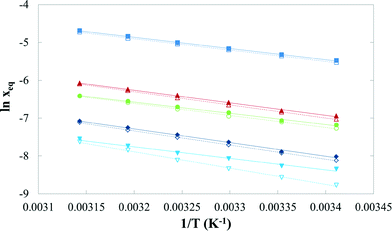 |
| | Fig. 17 lnxeq of quinizarin FI (solid symbols) and FII (hollow symbols) vs. 1/T in: ■, toluene; ▲, acetone; ●, acetic acid; ◆, acetonitrile; and ▼, n-butanol. | |
The mole fraction solubility obtained at different temperatures in each solvent was correlated using eqn (1):
| | | lnxeq = A + B/T | (1) |
where
T is in units of K. The coefficients of
eqn (1) obtained for each solvent by least squares fitting to the mole fraction solubility data are displayed in
Table 4.
R2 values for all systems exceed 0.99.
Table 4 Solubility regression coefficients (eqn (1)) for FI and FII in five solvents in the T range of 20 °C to 45 °C
| Solvent |
FI |
FII |
|
A
|
B
|
A
|
B
|
| Acetic acid |
2.9110 |
−2964.6 |
3.6166 |
−3195.7 |
| Acetone |
4.3868 |
−3327.4 |
4.8512 |
−3484.5 |
| Acetonitrile |
4.2780 |
−3613.4 |
4.6314 |
−3740.3 |
|
n-Butanol |
1.9620 |
−3037.6 |
5.9597 |
−4327.7 |
| Toluene |
4.6154 |
−2961.9 |
4.6699 |
−2990.4 |
The apparent or van't Hoff enthalpy of solution38 is essentially constant over the temperature range studied:
| | 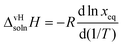 | (2) |
where
R is the universal gas constant and the derivative is the slope of each system obtained from the van't Hoff plot (
Fig. 17),
i.e., the regression coefficients
B in
Table 4. The values of
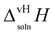
calculated for the two polymorphs in five solvents are given in
Table 5.
Table 5 van't Hoff enthalpy of solution of quinizarin (FI and FII) in five solvents from 20 °C to 45 °C
| Solvent |
ΔvHsolnH (kJ mol−1) |
| FI |
FII |
| Acetic acid |
24.6 |
26.6 |
| Acetone |
27.7 |
29.0 |
| Acetonitrile |
30.0 |
31.1 |
|
n-Butanol |
25.3 |
36.0 |
| Toluene |
24.6 |
24.9 |
Overall, mainly as a consequence of the high stability of the solid phase over the investigated temperature interval, the solubility is quite low. The highest solubility, expressed both as mole and mass fractions, is found in toluene, followed by acetone, acetic acid, acetonitrile and n-butanol. The solubility of FII is lower than FI, reaffirming the higher stability of FII over this temperature range. However, careful examination of the data for the two forms in each solvent indicates that a transition in thermodynamic stability will occur not far above the experimental T-range. If eqn (1) and the coefficients in Table 4 are used for extrapolation of the solubility data, on average over four solvents, acetic acid, acetone, acetonitrile and n-butanol, the estimated transition temperature is 64 °C. However, the value obtained from toluene (249 °C) is quite different from the others, which may be attributed partly to an increased uncertainty due to the relatively close, parallel solubility curves of FI and FII in this solvent.
In slurry conversion experiments performed between 30 °C and 80 °C in the five chosen solvents, the enantiotropic transition temperature was established to be between 50 °C and 60 °C, below which FII is the stable form. The rate of transformation is low in the vicinity of the transition temperature, but it could be established that over a period of three weeks a mixture of FI and FII transformed into FII at 50 °C and into FI at 60 °C. However, in the absence of FII crystals, the commercial FI exhibited sufficient kinetic stability to retain its structure over ten days in each of the five evaluated solvents at three temperatures (25 °C, 35 °C and 50 °C).
Unfortunately, solubility data above the estimated transition temperature could not be collected. However, assuming that the linear relationship between solubility and temperature can be extrapolated up to the transition point, an estimate of the enthalpy and entropy of transition can be obtained, e.g. according to the method outlined by Svärd et al.,39 by neglecting the influence of the activity coefficient on the ratio of solid state activities. The entropy of transformation can be expressed as:
| | 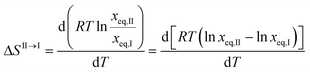 | (3) |
where
R is the universal gas constant,
T is the temperature in K, and the subscripts correspond to polymorphs I and II. Inserting
eqn (1), we get the following expression for the entropy of transformation at the transition temperature,
Ttr:
| | 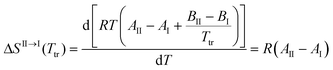 | (4) |
Eqn (4) shows that the entropy of transformation is independent of temperature over the range where solubility curves form linear van't Hoff plots. At the transition temperature, the Gibbs energy difference between the polymorphs is zero. With the calculated entropy values, the enthalpy difference can be estimated using eqn (5). The resulting data obtained in four solvents, using the transition temperatures obtained by extrapolation in each solvent, are presented in Table 6. Toluene was excluded from the analysis for previously outlined reasons.
| | | ΔHII→I(Ttr) = TtrΔSII→I = RTtr(AII − AI) | (5) |
Table 6 Entropy and enthalpy of transition at Ttr as estimated from solubility data in four solvents
| Solvent |
T
tr (°C) |
ΔHII→I (kJ mol−1) |
ΔSII→I (J mol−1 K−1) |
| Acetic acid |
55 |
1.92 |
5.87 |
| Acetone |
65 |
1.31 |
3.86 |
| Acetonitrile |
87 |
1.06 |
2.94 |
|
n-Butanol |
49 |
10.70 |
33.24 |
The calculations assume that the solubility ratio can approximate the corresponding activity ratio in all the solvents and that the temperature derivative of the saturated solution activity coefficients can likewise be neglected. The lack of consistency in the data obtained in the five solvents suggests that these assumptions are not well met, especially not in butanol.
The quinizarin molecule contains the fully conjugated cyclic dione structure embedded as in the 9,10-anthraquinone structure. The two ketone groups are, in principle, hydrogen bond accepting. In quinizarin, there are, in addition to the anthraquinone structure, two alcohol groups in para-position, and these have hydrogen bond accepting as well as donating functionality. However, the proximity of each ketone group with each respective alcohol group leads to intra-molecular hydrogen bonding, reducing the inter-molecular hydrogen bond accepting functionality of the ketone groups and the hydrogen bond donating capability of the alcohol groups. Obviously, solvation of the hydrophobic parts of the molecule plays an important role for the solubility and should explain why the solubility is highest in toluene. The results further show that the hydrogen bonding of acetone and acetonitrile to the alcohol groups of quinizarin does not sufficiently compensate for the lower capability of these solvents of solvating the hydrophobic part. n-Butanol is able to hydrogen bond to the ketones as well as to the alcohol groups, but in spite of this, the solubility in this solvent is the lowest. The acetic acid solubility is at first surprising. Of course, acetic acid has the capability to hydrogen bond to both the ketone groups and the alcohol groups, but in fact the strongest bonding is expected to occur as dimerization of two acetic acid molecules. Perhaps this dimerization actually provides acetic acid with a greater capability to solvate the hydrophobic parts of the quinizarin molecule. Single acetic acid molecules can hydrogen bond to the polar parts of the molecule, while dimers can solvate the hydrophobic parts.
Conclusions
The crystal structure of FII has been solved and a high-temperature polymorph of quinizarin (FIII) has been detected using DSC and HT-XRD. An attempt to index FIII PXRD data at 180 °C yielded a triclinic P cell and a Le Bail analysis confirmed the match of the cell to experimental data. The influence of solvent on crystal morphology and polymorphic form has been investigated by slow evaporation and cooling experiments, and there are no distinguishing shape differences between FI and FII. The solubility of quinizarin FI and FII in organic solvents decreases in the order: toluene, acetone, acetic acid, acetonitrile and n-butanol while no solvates were found. The solubility is overall low, below 2.5% by weight, over the range of conditions of this work. All van't Hoff curves are approximately linear over the investigated temperature interval. There is an enantiotropic relationship between FI and FII with a transition temperature experimentally determined to be between 50 °C and 60 °C. The commercial solid FI is found to be metastable in relation to FII below the transition temperature. However, at ambient temperature and in the five solvents investigated, the transformation in the absence of FII is slow, requiring many days.
Acknowledgements
The financial support of the Science Foundation Ireland (10/IN.1/B3038) and of the Swedish Research Council (621-2010-5391) is gratefully acknowledged.
References
-
H.-H. Tung, E. L. Paul, M. Midler and J. A. McCauley, Crystallization of organic compounds: an industrial perspective, John Wiley & Sons, 2009 Search PubMed.
-
A. G. Jones, Crystallization process systems, Butterworth-Heinemann, 2002, ch. 3 Search PubMed.
-
X. Z. Wang, C. Ma and K. J. Roberts, in Computer Aided Chemical Engineering, ed. B. Bertrand and J. Xavier, Elsevier, 2008, vol. 25, pp. 817–822 Search PubMed.
-
J. Bernstein, in Polymorphism: in the pharmaceutical industry, ed. R. Hilfiker, John Wiley & Sons, 2006, ch. 14 Search PubMed.
- Q. Huang, G. Lu, H. M. Shen, M. Chung and C. N. Ong, Med. Res. Rev., 2006, 27, 609–630 CrossRef PubMed.
- M. Locatelli, F. Tammaro, L. Menghini, G. Carlucci, F. Epifano and S. Genovese, Phytochem. Lett., 2009, 2, 223–226 CrossRef CAS PubMed.
- D. Jacquemin, J. Preat, V. Wathelet, J.-M. André and E. A. Perpète, Chem. Phys. Lett., 2005, 405, 429–433 CrossRef CAS PubMed.
- H. Oshio and N. Kawamura, Shoyakugaku Zasshi, 1985, 39, 131–138 CAS.
- X. Zhou and Q. Chen, Yaoxue Xuebao, 1988, 23, 17–20 CAS.
- G.-C. Yen, P.-D. Duh and D.-Y. Chuang, Food Chem., 2000, 70, 437–441 CrossRef CAS.
- S. K. Agarwal, S. S. Singh, S. Verma and S. Kumar, J. Ethnopharmacol., 2000, 72, 43–46 CrossRef CAS.
- A. Ali, N. Ismail, M. Mackeen, L. Yazan, S. Mohamed, A. Ho and N. Lajis, Pharm. Biol., 2000, 38, 298–301 CrossRef CAS.
- G. E. Xie, X. Zhu, Q. Li, M. Gu, Z. He, J. Wu, J. Li, Y. Lin, M. Li and Z. She, Br. J. Pharmacol., 2010, 159, 689–697 CrossRef CAS PubMed.
- J.-H. Chiang, J.-S. Yang, C.-Y. Ma, M.-D. Yang, H.-Y. Huang, T.-C. Hsia, H.-M. Kuo, P.-P. Wu, T.-H. Lee and J.-G. Chung, Chem. Res. Toxicol., 2010, 24, 20–29 CrossRef PubMed.
-
R. H. Chung, in Kirk-Othmer Encyclopedia of Chemical Technology, ed. M. Grayson and D. Eckroth, Wiley, New York, 3rd edn, 1978, vol. 2, pp. 708–757 Search PubMed.
-
H. Zollinger, Color chemistry: syntheses, properties, and applications of organic dyes and pigments, Wiley-VCH, 2004 Search PubMed.
-
L. L. Brunton, J. S. Lazo and K. L. Parker, Goodman & Gilman's the pharmacological basis of therapeutics, 2006 Search PubMed.
- R. S. Bottei and D. A. Lusardi, Thermochim. Acta, 1981, 43, 355–363 CrossRef CAS.
- S. Rossi, C. Tabolacci, A. Lentini, B. Provenzano, F. Carlomosti, S. Frezzotti and S. Beninati, Anticancer Res., 2010, 30, 445–449 CAS.
- P. K. Dutta and B. Lee, J. Raman Spectrosc., 1988, 19, 175–178 CrossRef CAS PubMed.
- O. Borgen, Acta Chem. Scand., 1966, 20, 2885–2886 CrossRef CAS PubMed.
- S. Swaminathan and G. D. Nigam, Curr. Sci., 1967, 36, 541 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 64, 112–122 CrossRef PubMed.
- P. McArdle, K. Gilligan, D. Cunningham, R. Dark and M. Mahon, CrystEngComm, 2004, 6, 303–309 RSC.
- G. C. Feast, J. Haestier, L. W. Page, J. Robertson, A. L. Thompson and D. J. Watkin, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, o635–o638 CAS.
- G. Smulevich, L. Angeloni, S. Giovannardi and M. P. Marzocchi, Chem. Phys., 1982, 65, 313–322 CrossRef CAS.
- J. Bernede, T. Ben Nasrallah, M. Jamali, J. Mevellec, C. Rabiller and A. Proutiere, J. Phys. Chem. Solids, 1995, 56, 1239–1251 CrossRef CAS.
- T. B. Nasrallah, J. C. Bernede, A. Godoy, C. Rabiller and D. Legoff, J. Solid State Chem., 1993, 106, 388–399 CrossRef.
- X. Xuan, X. Wang and N. Wang, Spectrochim. Acta, Part A, 2011, 79, 1091–1098 CrossRef CAS PubMed.
-
A. Larson and R. Von Dreele, Los Alamos National Laboratory, document LAUR, 1987, pp. 86–748 Search PubMed.
- B. H. Toby, J. Appl. Crystallogr., 2005, 38, 1040–1041 CrossRef CAS.
- J. Visser, J. Appl. Crystallogr., 1969, 2, 89–95 CrossRef CAS.
- P. Hartman and W. Perdok, Acta Crystallogr., Sect. A: Found. Adv., 1955, 8, 49–52 Search PubMed.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef CAS.
- P. McArdle, Y. Hu, A. Lyons and R. Dark, CrystEngComm, 2010, 12, 3119–3125 RSC.
- N. Panina, R. Van de Ven, F. Janssen, H. Meekes, E. Vlieg and G. Deroover, Cryst. Growth Des., 2008, 9, 840–847 Search PubMed.
- J. Karpinska, A. Erxleben and P. McArdle, Cryst. Growth Des., 2013, 13, 1122–1130 CAS.
- F. L. Nordström and Å. C. Rasmuson, Eur. J. Pharm. Sci., 2009, 36, 330–344 CrossRef PubMed.
- M. Svärd, F. L. Nordström, E.-M. Hoffmann, B. Aziz and Å. C. Rasmuson, CrystEngComm, 2013, 15, 5020–5031 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra of FI and FII, crystallographic data of quinizarin FI and FII, hydrogen-bond geometry in the crystal structures of quinizarin FI and FII. CCDC 1044980 and 1044981. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce00147a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Colin
Seaton
c,
Patrick
McArdle
d and
Åke C.
Rasmuson
*ab
ab,
Colin
Seaton
c,
Patrick
McArdle
d and
Åke C.
Rasmuson
*ab




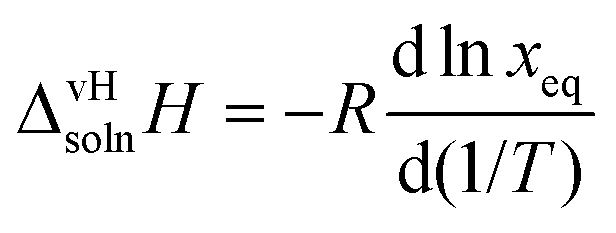
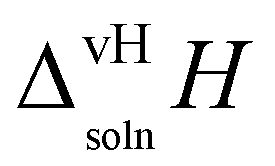 calculated for the two polymorphs in five solvents are given in Table 5.
calculated for the two polymorphs in five solvents are given in Table 5.