
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChirality in charge-transfer salts of BEDT-TTF of tris(oxalato)chromate(III)†
Lee
Martin
*a,
Hiroki
Akutsu
bc,
Peter N.
Horton
d and
Michael B.
Hursthouse
d
aSchool of Science and Technology, Nottingham Trent University, Clifton Lane, Clifton, Nottingham, NG11 8NS, UK. E-mail: lee.martin@ntu.ac.uk; Fax: +44 (0)1158488077; Tel: +44 (0)1158483128
bDepartment of Chemistry, Graduate School of Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan
cGraduate School of Material Science, University of Hyogo, Kamigori-cho, Ako-gun, Hyogo, 678-1297, Japan
dSchool of Chemistry, Faculty of Natural and Environmental Sciences, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
First published on 13th February 2015
Abstract
Crystallisation from chiral electrolyte (R)-(−)-carvone has produced three new chiral semiconducting salts of BEDT-TTF from racemic anion tris(oxalato)chromate(III).
Introduction
Radical cation salts of bis(ethylenedithio)tetrathiafulvane (BEDT-TTF) and its derivatives have been extensively studied owing to their ability to combine physical properties through crystal engineering of the cationic and anionic layers. BEDT-TTF salts containing tris(oxalato)metallate anions have provided a large family of materials of the formula (BEDT-TTF)4[(A)M3+(C2O4)3]·guest (A = K+, H3O+, NH4+; M3+ = Fe, Cr, Ga, Al, Co, Ru) which combine paramagnetism and superconductivity in the same lattice.1 The tris(oxalato)metallate anion layers form a honeycomb-like network with a hexagonal cavity capable of including a guest solvent molecule from the electrolyte being used for electrochemical synthesis of the material. Changing the shape and size of the guest solvent molecule within the anion layer causes subtle changes in the hydrogen bonding interactions between insulating anion layer and conducting cation layer and leads to a variety of ground states. β′′ donor packing is observed in many cases and depending on the guest solvent molecule leads to superconductivity (M = Fe3+, guest = PhCN, PhNO2, PhBr, PhF),1,2 metallic (M = Fe3+, guest = PhCl, DMF),3 semiconducting (M = Fe3+, guest = PhI)4 or metal–insulator behaviour (M = Fe3+, guest = pyridine).5 In some cases the electrolyte molecule is not included in the hexagonal cavity due to its size/shape e.g. 1,2,4-trichlorobenzene,6 carvone.7 However it has been shown that 1,2,4-trichlorobenzene is an excellent solvent in which to grow crystals of this type in combination with a smaller guest molecule and ethanol.6When a guest solvent molecule is slightly too large to fit in the cavity it protrudes from one side of the anion layer and results in the two faces of the anion layer having different interactions with the neighbouring donor layers. This gives rise to two different donor packing types with different electronic ground states in alternate layers. α-Pseudo-κ packing has been found with 1,2-dibromobenzene giving metallic behaviour,8 whilst a metal–insulator transition is observed in the α–β′′ phases obtained from PhN(CH3)COH, PhCH2CN or PhCOCH3.9 An α–β′′ phase is also obtained when the guest molecule is racemic (R/S)- or chiral (S)-sec-phenethyl alcohol.10 The two salts formed are isostructural, however, small differences in the metal-insulating properties are observed owing to the enantiomeric disorder observed in the (R/S)-salt versus the (S)-salt.
The incorporation of chirality into this family of salts has the potential to introduce new properties such as the Hall effect.11 Rikken has also observed magneto-chiral anisotropy in carbon nanotubes,12 where the resistivity along nanotubes with opposite chirality are different in a coaxial magnetic field. Chirality can be introduced into radical salts using a chiral donor molecule, chiral anion, or through the use of a chiral electrolyte as the guest molecule.
Since the first enantiopure TTF-based donor molecule tetramethyl-(S,S,S,S)-BEDT-TTF,13 there have been a large number of chiral donor molecules synthesized.14 Recently, a novel TTF donor has shown lower activation energy for its racemic salt with BF4 compared to the isostructural analogues for both individual enantiomers.15 The great promise of this area of research is shown by the observation of electrical magnetochiral anisotropy in enantiopure (DM-EDT-TTF)2ClO4.16
BEDT-TTF has been used as the donor with a variety of racemic/chiral anions such as MIII(oxalate)3,1–10 Fe(croconate)3,17 Cr(2,2′-bipy)(oxalate)2,18 Sb2(L-tartrate)2,19 TRISPHAT20 and Fe(C6O4Cl2)3.21
We have tried to take advantage of the chirality of the tris(oxalato)metallate anion to produce an overall chiral crystal of the superconducting β′′-(BEDT-TTF)4[(H3O)Cr(C2O4)3]·C6H5CN in which each anion layer contains only a single enantiomer of Cr(oxalate)3.22 Neighbouring anion layers contain only the opposing enantiomer giving a Δ–Λ–Δ–Λ–Δ–Λ–Δ–Λ repeating pattern (where – represents a BEDT-TTF layer). When starting from enantiomerically pure tris(oxalato)metallates they racemise rapidly in solution under the conditions of crystal growth also leading to an overall racemic lattice but with a different spatial distribution of the enantiomers compared to the β′′ phase.22 The polymorph grown from enantiopure Cr(oxalate)3 has a semiconducting pseudo-κ donor arrangement in which every anion layer contains a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mix of both Δ and Λ enantiomers in alternating rows giving a ΔΛ–ΔΛ–ΔΛ–ΔΛ pattern.
:50 mix of both Δ and Λ enantiomers in alternating rows giving a ΔΛ–ΔΛ–ΔΛ–ΔΛ pattern.
We have successfully synthesised the first chiral examples in this large family of radical cation salts containing M(oxalate)3 by using chiral induction. Through electrocrystallisation from chiral electrolyte (R)-(−)-carvone containing racemic Cr(C2O4)3 we obtained semiconducting (BEDT-TTF)3NaCr(C2O4)3·guest (guest = CH2Cl2 or CH3NO2).7 We report here three new additions to this family by introducing new guest molecules into the hexagonal cavity within the honeycomb network of the anion layer created by Na and Cr(C2O4)3.
Using DMF (I), acetonitrile (II), or ethanol (III) 50:50 with (R)-(−)-carvone as supporting electrolyte in the electrocrystallisation of Na3Cr(C2O4)3 and 15-crown-5 with BEDT-TTF yields chiral crystals of the form (BEDT-TTF)3NaCr(C2O4)3·guest.
Salts I–III all consist of alternate anionic and cationic layers. The anionic layers contain a honeycomb network of Na+ and Cr(oxalate)33− with a solvent molecule sited in the hexagonal cavity. Na⋯Cr distances of 5.35(1), 5.72(1) and 5.76(1) Å for I, 5.49(1), 5.54(1) and 5.62(1) Å for II, and 5.52(1), 5.54(1) and 5.59(1) Å for III, are shorter than the O⋯Cr or K⋯Cr distances found in the β′′ superconducting phases where H3O+, NH4+ or K+ are the counter cation. The resulting hexagonal cavities are therefore smaller in size in these Na salts and smaller guest molecules are preferred in these salts (nitromethane, dichloromethane, DMF, ethanol, acetonitrile)7 compared to the β′′ phases (e.g. benzonitrile, chlorobenzene).1–5
Salts I–III each have a different BEDT-TTF donor packing motif. When the guest molecule is DMF the donors have a θ-type packing motif, whilst when the guest is acetonitrile or ethanol the packing is isostructural to that observed previously when the guest is nitromethane or dichloromethane, respectively.7
Results and discussion
θ-(BEDT-TTF)3NaCr(C2O4)3·DMF, I, (Fig. 1) crystallises in space group P1. The inclusion of (R)-(−)-carvone in the synthesis of I has the effect of producing a different phase to that produced when dimethylformamide is used alone. When DMF is used as the sole electrolyte it produces a β′′ phase (BEDT-TTF)4[(A)M3+(C2O4)3]·DMF (A = K+, NH4+; M3+ = Fe, Cr) which is metallic down to 4.2 K.23 In the β′′ phase the DMF molecule is located on a two-fold symmetry axis and exhibits disorder of the position of the methyl groups and the carbon atom, with the O atom pointing towards a metal centre. In I the inorganic layer also adopts a hexagonal packing of trisoxalates but with only a single enantiomer of Cr(C2O4)33− present. The crystal used is a non-merohedral twin (35%), and to model this produces an unreliable Flack parameter (−0.4(2)). This along with disorder between the Na and Cr sites indicates that the crystal is an inversion twin. Due to this twinning and disorder it is difficult to deconvalute the overall architecture, so that although the only independent DMF molecule in I sits in the hexagonal cavity the Na and Cr disorder means it can exist disordered over 2 sites with respect to the layer. However it only ever sticks out to one side of layer (Fig. 1 and 2). | ||
| Fig. 1 Layered structure of I. Hydrogens are omitted for clarity. The anion layer is heavily disordered so only D and F are shown (see Fig. 2 for explanation of symbols). | ||
 | ||
| Fig. 2 Anion layer of I viewed down the c axis. Hydrogens are omitted for clarity. Owing to the short distance of the a-axis of only 5.3536(5) Å the molecular size of tris(oxalato)metallate is larger than the a-axis. The anion layer is disordered and thus is represented by the four separate figures (a)–(d), whilst the true anion layer can be constructed by superimposing all four figures. There are two crystallographically independent tris(oxalato)metallates (C and D) and two DMF molecules (E and F). In (a) only C and E are described. Four tris(oxalato)metallates in (a) are C(x, y, z), C(x, y − 1, z), C(x − 2, y, z) and C(x − 2, y − 1, z) where symmetry codes are indicated in the parentheses. We did not draw the molecules of C(x − 1, y, z) and C(x − 1, y − 1, z) in (a), which were drawn in (b) with additional molecules of C(x + 1, y, z) and C(x + 1, y − 1, z) because molecules in (a) and (b) are overlapped to be complicated. Figures (c) and (d) are similar figures for D and F. Each DMF molecule is located at almost the same position of a centre of tris(oxalato)metallate. We assume that the structure is three-fold (see text for details). Figure (e) is the estimated ordered structure of the anion layer where the a-axis is three times the original a-axis. | ||
Unique in this family of BEDT-TTF with tris(oxalato)metallate the donors have θ packing (Fig. 3). One BEDT-TTF molecule has disordered ethylene groups at both ends whilst the other has disorder at one end only. Table 1 estimates charges for the two independent BEDT-TTF molecules based on their C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–S bond distances. Donor A within one stack each have a charge of 0.99+ with donor B in the other stack having charges of 0.46+. This is consistent with the formula of two BEDT-TTFs of 1.45+ ± 0.2 per two-thirds of a NaCr(C2O4)3 having a charge of 1.33−.
C and C–S bond distances. Donor A within one stack each have a charge of 0.99+ with donor B in the other stack having charges of 0.46+. This is consistent with the formula of two BEDT-TTFs of 1.45+ ± 0.2 per two-thirds of a NaCr(C2O4)3 having a charge of 1.33−.
 | ||
| Fig. 3 BEDT-TTF layer of I viewed down the c axis showing S⋯S contacts below the sum of the van der Waals radii (<3.7 Å). It has an θ-type packing motif. The dihedral angle between A and B molecules of 111.9° suggests that the ground state is a non-magnetic insulator according to the θ-phase diagram (so called ‘Mori diagram’ H. Mori, J. Phys. Soc. Jpn., 2006, 75(5), 051003). | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Salt | Donor | a/Å | b/Å | c/Å | d/Å | δ | Q | Charge |
| I | A | 1.411 | 1.732 | 1.744 | 1.347 | 0.718 | 0.99+ | |
| B | 1.366 | 1.733 | 1.758 | 1.336 | 0.789 | 0.46+ | 1.45 ± 0.2 = 1.33 | |
| II | C | 1.350 | 1.756 | 1.759 | 1.338 | 0.827 | 0.18+ | |
| B | 1.390 | 1.731 | 1.756 | 1.302 | 0.795 | 0.41+ | ||
| A | 1.418 | 1.725 | 1.739 | 1.384 | 0.662 | 1.41+ | 2.00 ± 0.3 = 2 | |
| III | C | 1.361 | 1.748 | 1.755 | 1.363 | 0.779 | 0.53+ | |
| B | 1.376 | 1.733 | 1.749 | 1.356 | 0.750 | 0.75+ | ||
| A | 1.373 | 1.737 | 1.751 | 1.349 | 0.766 | 0.63+ | 1.91 ± 0.3 = 2 | |

Within the BEDT-TTF stacks there are close S⋯S contacts below the sum of the van der Waals radii (<3.6 Å) S2⋯S12 3.44(1), S6⋯S17 3.45(1), S7⋯S17 3.43(1), S1⋯S11 3.47(1), S4⋯S11 3.45(1), S8⋯S18 3.47(1) Å. There are also several side-to-side contacts between the stacks S7⋯S16 3.44(1), S7⋯S17 3.42(1), S2⋯S12 3.57(1), S1⋯S11 3.44(1), S1⋯S14 3.45(1) Å. Two-probe resistivity measurement on a single crystal show that I is a semiconductor with an activation energy of 0.043 eV, with a room temperature resistivity ρRT of 0.26 Ohm cm.
The anionic layer is highly disordered and the structure is complicated, therefore we use four separate figures as shown in Fig. 2(a)–(d). There are two crystallographically independent donor molecules, two NaCr(C2O4)3 and two DMF molecules. The cell volume being only 929.12(14) Å3, we determined that all of the occupancies of NaCr(C2O4)3 and DMF are 1/3. Therefore, the composition is (BEDT-TTF)2[NaCr(C2O4)3]2/3·(DMF)2/3. Because of the composition, two thirds of the oxalate anion can be occupied in the unit cell. In other words, C(x, y, z) and C(x − 1, y, z) are not occupied at the same time due to the short a-axis of 5.3536(5) Å. The lattice has to have three-fold periodicity along the a-axis to attain the two thirds filling. Therefore, three unit cells of the anion layer consist of two NaCr(C2O4)3 and two DMF molecules. We assume three-fold periodicity along the a-axis and the estimated ordered structure is shown in Fig. 2(e). If the hypothesis is true, the crystal should show extra diffuse streaks or satellite spots. Some photographs appear to have some diffuse streaks along the a-axis on h0l planes. However, because of the small crystal size, the intensities are almost the same as those of the relatively large background.
(BEDT-TTF)3NaCr(C2O4)3·CH3CN, II, (Fig. 4) crystallises in monoclinic space group P21. Acetonitrile has previously been used as electrolyte with BEDT-TTF and M(C2O4)3 to synthesise salts in this family25 but a β′′ phase has not been obtained. The inclusion of (R)-(−)-carvone with acetonitrile in the synthesis of II has the effect of producing a new phase containing only a single enantiomer of Cr(C2O4)33−. The structure of compound II is isostructural with the salt reported previously when using nitromethane/(R)-(−)-carvone as the electrolyte. The asymmetric unit of II contains three crystallographically independent BEDT-TTFs, two are parallel to one another (A and B) with third molecule (C) at 45° with respect to the other two (Fig. 5). There are no face-to-face close S⋯S contacts between the BEDT-TTFs A and B, but there are two side-to-side close contacts (S7⋯S17 3.56(1) and S6⋯S17 3.56(1) Å). The shortest of the S⋯S contacts are between donor C and either A or B, S1⋯S27 3.58(1), S8⋯S23 3.51(1), S8⋯S22 3.48(1), S17⋯S25 3.53(1), and S17⋯S28 3.54(1) Å. There are no short contacts between adjacent C BEDT-TTFs (Fig. 5).
 | ||
| Fig. 4 Layered structure of II showing atomic labels. Hydrogens are omitted for clarity. | ||
 | ||
| Fig. 5 BEDT-TTF layer of II viewed down the molecular long axis showing S⋯S contacts below the sum of the van der Waals radii (<3.7 Å). Hydrogens are omitted for clarity. | ||
The anion layer of II contains only a single enantiomer of Cr(C2O4)33−. However because of the relatively poor quality of crystal data (high R-value of 9.9% and with a Flack parameter of 0.27(12)), we cannot state whether it is enantiopure or not, nor whether salt II is an inversion twin. The acetonitrile molecule is sited with the hexagonal cavity created by Na and Cr(C2O4)3 with the acetonitrile nitrogen atom protruding out on opposite sides of the anion layer on alternating sides in adjacent rows (Fig. 4 and 6). This nitrogen atom has a number of close contacts involving all three of the crystallographically independent BEDT-TTF molecules N61⋯H1B 2.48(1), N61⋯C1 3.17(1), N61⋯S1 3.17(1), N61⋯C20 3.02(1), N61⋯H20F 2.27(1) and N61⋯H29A 2.67(1) Å.
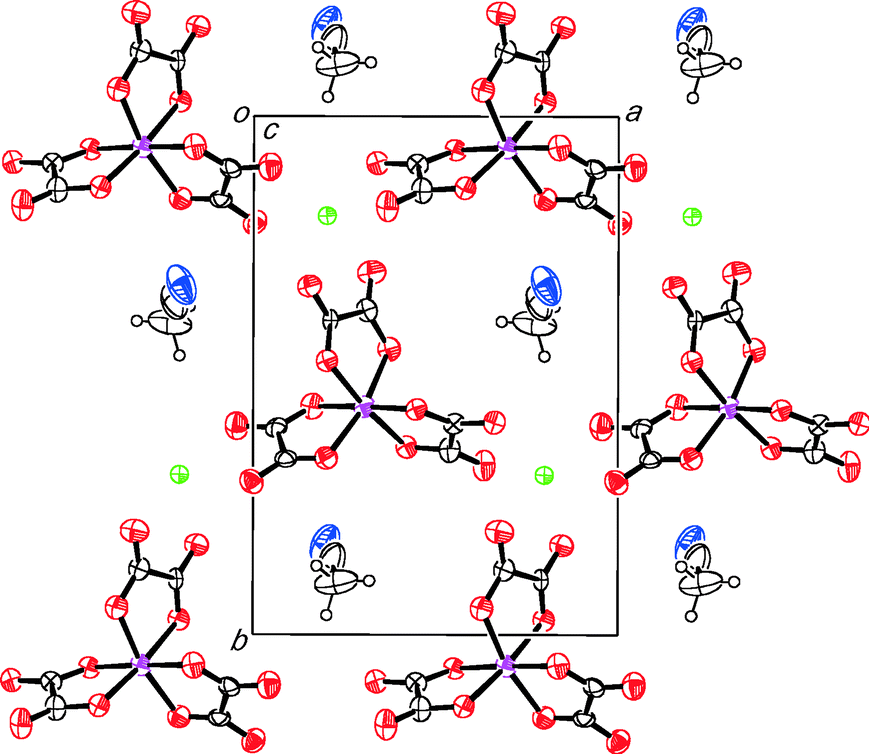 | ||
| Fig. 6 Anion layer of II viewed down the c axis. | ||
Based on their CC and C–S bond distances24 (Table 1) the three crystallographically independent BEDT-TTF molecules C, B and A have charges of 0.18+, 0.41+, and 1.41+, respectively. This is consistent with the formula of three BEDT-TTFs of 2.00+ ± 0.3 per [NaCr(C2O4)3]2−. A molecule having a charge of more than +1 is somewhat unusual,26 which may be due to the relatively high R-value. The charge of donor C is almost zero and the total charge of A and B donors are approximately +2, suggesting that the salt is in a charge disproportionation state where the monomer C is neutral and the dimer A–B has two holes which forms a spin dimer. These donor charges show a significant difference compared to the isostructural nitromethane salt (BEDT-TTF)3NaCr(C2O4)3·CH3NO2 where charges are calculated as 0.50+, 0.64+, and 0.69+. Giving a total of 1.85+ ± 0.3 suggesting that the holes on the donor layer are more delocalized in this case. Two-probe resistivity measurements were made on a single crystal and show that II is a semiconductor with an activation energy of 0.172 eV and a room temperature resistivity of ρRT of 26.5 Ohm cm. The ρRT value is similar to that of the nitromethane salt (22 Ohm cm) but the activation energy is higher than that of the nitromethane salt (0.079 eV).
(BEDT-TTF)3NaCr(C2O4)3·CH3OH, III, (Fig. 7–9) crystallises in space group P1. Ethanol is often added to aid the dissolution of M(C2O4)3 in the electrolyte when synthesizing β′′ phase salts using K+ or NH4+ as the counter cation,6 however the ethanol is not included in the structure. Zhang et al.27 have synthesised an antiferromagnetic semiconductor from BEDT-TTF and (Et3NH)2Cu(C2O4)2 which includes two ethanol molecules in each hexagonal cavity created by six oxalate-bridged copper stoms. The use of sodium tris(oxalato)metallate with (R)-(−)-carvone and ethanol in the synthesis of III has the effect of producing a new phase with a honeycomb arrangement of Na and a single enantiomer of Cr(C2O4)33− with ethanol in the hexagonal cavity.
 | ||
| Fig. 7 Layered structure of III showing atomic labels. Hydrogens are omitted for clarity. | ||
 | ||
| Fig. 8 BEDT-TTF layer of III viewed down the molecular long axis showing S⋯S contacts below the sum of the van der Waals radii (<3.7 Å). Hydrogens are omitted for clarity. | ||
 | ||
| Fig. 9 Anion layer of I viewed down the c axis. Hydrogens are omitted for clarity. | ||
The structure of compound III is isostructural with the salt (BEDT-TTF)3NaCr(C2O4)3·DCM reported previously.7 The asymmetric unit of III consists of three crystallographically independent BEDT-TTF molecules (Fig. 8). Two of the molecules are face-to-face with the third molecule at 80° with respect to the others. There is only one side-to-side S⋯S contacts between the face-to-face pair of BEDT-TTFs, S1⋯S28 3.57(1) Å. The shorter S⋯S contacts are all between the third BEDT-TTF and one or other molecules of the face-to-face pair S6⋯S11 3.42(1), S7⋯S11 3.49(1), S2⋯S15 3.48(1), S2⋯S18 3.56(1), S12⋯S22 3.53(1), S13⋯S22 3.53(1), S17⋯S26 3.43(1), S15⋯S28 3.56(1) and S17⋯S27 3.50(1) Å (Fig. 8).
The anion layer of III contains only a single enantiomer of Cr(C2O4)33− with a Flack parameter of 0.51(2) indicating that salt III is an inversion twin. The ethanol molecule is sited within the hexagonal cavity created by Na and Cr(C2O4)3 and is disordered over two positions (Fig. 7 and 9).
Based on their CC and C–S bond distances24 (Table 1) the three crystallographically independent BEDT-TTF molecules C, B and A have charges of 0.53+, 0.75+, and 0.63+, respectively This is consistent with the formula of three BEDT-TTFs of 1.91+ ± 0.3 per [NaCr(C2O4)3]2−. In this salt the charges on the ET layer are delocalized similar to the nitromethane salt, suggesting lower resistivity and activation energy than II. However, crystals of III were too small and fragile for measurement of resistivity.
Experimental
Synthesis and purification of starting materials
Na3Cr(C2O4)3 was synthesised by an adaptation of the method of Bailar and Jones.28 BEDT-TTF was purchased from Sigma Aldrich and recrystallised from chloroform. Ethanol, DMF, acetonitrile and 15-crown-5 were purchased from Sigma Aldrich and used as received.Synthesis of charge-transfer salts
Crystals of I–III were grown on platinum electrodes by electrocrystallisation in 40 ml H-shaped electrochemical cells.‡Platinum electrodes were cleaned by applying a voltage across the electrodes in 1 M H2SO4 in each direction to produce H2 and O2 at the electrodes, then washed with distilled water and thoroughly dried.
Na3[Cr(C2O4)3]·3H2O (120 mg) and 15-crown-5 (200 mg) were dissolved in a mixture of 20 ml (R)-(−)-carvone and 20 ml of a 2nd solvent (DMF for I, acetonitrile for II, or ethanol for III) with stirring overnight before filtering into an H-cell containing 10 mg BEDT-TTF in the anode compartment.
H-cells were placed in a dark box on a vibration-free bench at a constant current of 1.0 μA and after 2 weeks a large number of black crystals were harvested from the anode.
Physical measurements
Two-probe DC transport measurements were made on crystals using a HUSO HECS 994C multi-channel conductometer. Gold wires (15 μm diameter) were attached to the crystal, and the attached wires were connected to an integrated circuit plug with gold conductive cement.Conclusions
We have shown that it is possible to synthesise chiral molecular conductors using the readily available achiral BEDT-TTF donor molecule with inexpensive electrolytes and racemic anions. Three new charge-transfer salts of BEDT-TTF and [Cr(C2O4)3]3− have been synthesised using a chiral solvent to bring about chiral induction, i.e. crystallisation of a chiral salt from a racemic precursor.The hexagonal cavities are smaller in size in these Na salts and smaller guest molecules are preferred in these salts (nitromethane, dichloromethane, DMF, ethanol, acetonitrile)7 compared to the β′′ phases (e.g. benzonitrile, chlorobenzene). We are continuing to synthesise further salts of this type family using other solvents and anions to more closely examine the effect of chirality upon the physical properties.
Acknowledgements
LM thanks the Royal Society of Chemistry for a Journals Grant for International Authors. This work has been supported by the Royal Society [Research grants (RG100853 and RG081209), International Exchange Scheme (IE130367), and International Joint Project (JP0869972)]. We thank EPSRC for funding the National Crystallography Service.Notes and references
- M. Kurmoo, A. W. Graham, P. Day, S. J. Coles, M. B. Hursthouse, J. L. Caulfield, J. Singleton, F. L. Pratt, W. Hayes, L. Ducasse and P. Guionneau, J. Am. Chem. Soc., 1995, 117, 12209 CrossRef CAS.
- L. Martin, S. S. Turner, P. Day, P. Guionneau, J. K. Howard, D. E. Hibbs, M. E. Light, M. B. Hursthouse, M. Uruichi and K. Yakushi, Inorg. Chem., 2001, 40, 1363 CrossRef CAS PubMed; S. Rashid, S. S. Turner, P. Day, J. A. K. Howard, P. Guionneau, E. J. L. McInnes, F. E. Mabbs, R. J. H. Clark, S. Firth and T. Biggs, J. Mater. Chem., 2001, 11, 2095 RSC; E. Coronado, S. Curreli, C. Giménez-Saiz and C. J. Gómez-García, J. Mater. Chem., 2005, 15, 1429 RSC.
- T. G. Prokhorova, S. S. Khasanov, L. V. Zorina, L. I. Buravov, V. A. Tkacheva, A. A. Baskakov, R. B. Morgunov, M. Gener, E. Canadell, R. P. Shibaeva and E. B. Yagubskii, Adv. Funct. Mater., 2003, 13, 403 CrossRef CAS; E. Coronado, S. Curreli, C. Giménez-Saiz and C. J. Gómez-García, Synth. Met., 2005, 154, 245 CrossRef PubMed.
- E. Coronado, S. Curreli, C. Giménez-Saiz and C. J. Gómez-García, Inorg. Chem., 2012, 51, 1111 CrossRef CAS PubMed.
- S. S. Turner, P. Day, D. E. Hibbs, K. M. A. Malik, M. B. Hursthouse, S. J. Teat, E. J. MacLean, L. Martin and S. A. French, Inorg. Chem., 1999, 38, 3543 CrossRef CAS PubMed.
- T. G. Prokhorova, L. I. Buravov, E. B. Yagubskii, L. V. Zorina, S. S. Khasanov, S. V. Simonov, R. P. Shibaeva, A. V. Korobenko and V. N. Zverev, CrystEngComm, 2011, 13, 537 RSC.
- L. Martin, P. Day, P. Horton, S. Nakatsuji, J. Yamada and H. Akutsu, J. Mater. Chem., 2010, 20, 2738 RSC; L. Martin, P. Day, S. Nakatsuji, J. Yamada, H. Akutsu and P. Horton, CrystEngComm, 2010, 12, 1369 RSC.
- L. V. Zorina, S. S. Khasanov, S. V. Simonov, R. P. Shibaeva, V. N. Zverev, E. Canadell, T. G. Prokhorova and E. B. Yagubskii, CrystEngComm, 2011, 13, 2430 RSC.
- H. Akutsu, A. Akutsu-Sato, S. S. Turner, P. Day, E. Canadell, S. Firth, R. J. H. Clark, J.-I. Yamada and S.-I. Nakatsuji, Chem. Commun., 2004, 18 RSC.
- L. Martin, P. Day, H. Akutsu, J. Yamada, S. Nakatsuji, W. Clegg, R. W. Harrington, P. N. Horton, M. B. Hursthouse, P. McMillan and S. Firth, CrystEngComm, 2007, 9, 865 RSC.
- J. T. Chalker and S. L. Sondhi, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 4999 CrossRef CAS; A. Kleiner, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 155311 CrossRef; R. Roy and C. Kallin, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 174513 CrossRef.
- G. L. J. A. Rikken and E. Raupach, Nature, 1997, 390, 493 CrossRef CAS PubMed; G. L. J. A. Rikken, J. Folling and P. Wyder, Phys. Rev. Lett., 2001, 87, 236602 CrossRef; V. Krstic, S. Roth, M. Burghard, K. Kern and G. L. J. A. Rikken, J. Chem. Phys., 2002, 117, 11315 CrossRef PubMed; V. Krstic and G. L. J. A. Rikken, Chem. Phys. Lett., 2002, 364, 51 CrossRef; G. L. J. A. Rikken, Science, 2011, 331, 864 CrossRef PubMed.
- J. D. Dunitz, A. Karrer and J. D. Wallis, Helv. Chim. Acta, 1986, 69, 69 CrossRef.
- N. Avarvari and J. D. Wallis, J. Mater. Chem., 2009, 19, 4061 RSC; S. Yang, A. C. Brooks, L. Martin, P. Day, H. Li, P. Horton, L. Male and J. D. Wallis, CrystEngComm, 2009, 11, 993 RSC; J. D. Wallis and J. P. Griffiths, J. Mater. Chem., 2005, 15, 347 RSC; R. J. Brown, A. C. Brooks, J.-P. Griffiths, B. Vital, P. Day and J. D. Wallis, Org. Biomol. Chem., 2007, 3172 Search PubMed; C. Réthoré, N. Avarvari, E. Canadell, P. Auban-Senzier and M. Fourmigué, J. Am. Chem. Soc., 2005, 127, 5748 CrossRef CAS PubMed; M. Chas, M. Lemarié, M. Gulea and N. Avarvari, Chem. Commun., 2008, 220 RSC; F. Pop, P. Auban-Senzier, A. Frąckowiak, K. Ptaszyński, I. Olejniczak, J. D. Wallis, E. Canadell and N. Avarvari, J. Am. Chem. Soc., 2013, 135(45), 17176 CrossRef PubMed.
- L. Martin, J. D. Wallis, M. A. Guziak, J. Oxspring, J. R. Lopez, S.-I. Nakatsuji, J.-I. Yamada and H. Akutsu, CrystEngComm, 2014, 16, 5424 RSC; I. Awheda, S. Krivickas, S. Yang, L. Martin, M. A. Guziak, A. C. Brooks, F. Pelletier, M. Le Kerneau, P. Day, P. Horton, H. Akutsu and J. D. Wallis, Tetrahedron, 2013, 69, 8738 CrossRef CAS PubMed.
- F. Pop, P. Auban-Senzier, E. Canadell, G. L. J. A. Rikken and N. Avarvari, Nat. Commun., 2014, 5, 3757 Search PubMed.
- C. J. Gómez-García, E. Coronado, S. Curreli, C. Giménez-Saiz, P. Deplano, M. L. Mercuri, L. Pilia, A. Serpe, C. Faulmann and E. Canadell, Chem. Commun., 2006, 4931 RSC.
- A. M. Madalan, E. Canadell, P. Auban-Senzier, D. Brânzea, N. Avarvari and M. Andruh, New J. Chem., 2008, 32, 333 RSC.
- E. Coronado, J. R. Galan-Mascaros, C. J. Gomez-Garcia, A. Murcia-Martinez and E. Canadell, Inorg. Chem., 2004, 43, 8072 CrossRef CAS PubMed.
- M. Clemente-León, E. Coronado, C. J. Gómez-García, A. Soriano-Portillo, S. Constant, R. Frantz and J. Lacour, Inorg. Chim. Acta, 2007, 360, 955 CrossRef PubMed.
- S. Benmansour, E. Coronado, C. Giménez-Saiz, C. J. Gómez-García and C. Rößer, Eur. J. Inorg. Chem., 2014, 24, 3949 CrossRef; M. Atzor, F. Pop, P. Auban-Senzier, C. J. Gómez-García, E. Canadell, F. Artizzu, A. Serpe, P. Deplano, N. Avarvari and M. L. Mercuri, Inorg. Chem., 2014, 53(13), 7028 CrossRef PubMed.
- L. Martin, S. S. Turner, P. Day, F. E. Mabbs and E. J. L. McInnes, Chem. Commun., 1997, 1367 RSC; L. Martin, S. S. Turner, P. Day, K. M. A. Malik, S. J. Coles and M. B. Hursthouse, Chem. Commun., 1999, 513 RSC; L. Martin, S. S. Turner and P. Day, Synth. Met., 1999, 102, 1638 CrossRef CAS.
- T. G. Prokhorova, S. S. Khasanov, L. V. Zorina, L. I. Buravov, V. A. Tkacheva, A. A. Baskakov, R. B. Morgunov, M. Gener, E. Canadell, R. P. Shibaeva and E. B. Yagubskii, Adv. Funct. Mater., 2003, 13(5), 403 CrossRef CAS.
- P. Guionneau, C. J. Kepert, D. Chasseau, M. R. Truter and P. Day, Synth. Met., 1997, 86, 1973 CrossRef CAS.
- L. Martin, P. Day, A. Bingham, P. Horton and M. Hursthouse, J. Low Temp. Phys., 2006, 142, 417 CrossRef CAS; L. Martin, P. Day, W. Clegg, R. W. Harrington, P. N. Horton, A. Bingham, M. B. Hursthouse, P. McMillan and S. Firth, J. Mater. Chem., 2007, 31, 3324 RSC; L. Martin, P. Day, S. A. Barnett, D. A. Tocher, P. N. Horton and M. B. Hursthouse, CrystEngComm, 2008, 2, 192 RSC; L. Martin, P. Day, S.-I. Nakatsuji, J.-I. Yamada, H. Akutsu and P. Horton, CrystEngComm, 2010, 12, 1369 RSC.
- K. A. Abboud, M. B. Clevenger, G. F. de Oliveira and D. R. Talham, J. Chem. Soc., Chem. Commun., 1993, 1560 RSC; L.-K. Chou, M. A. Quijada, M. B. Clevenger, G. F. de Oliveira, K. A. Abboud, D. B. Tanner and D. R. Talham, Chem. Mater., 1995, 7, 530 CrossRef CAS.
- B. Zhang, Y. Zhang and D. Zhu, Chem. Commun., 2012, 48, 197 RSC.
- J. C. Bailar and E. M. Jones, Inorg. Synth., 1939, 1, 35 CrossRef.
Footnotes |
| † CCDC 1035041–1035043 contains supplementary X-ray crystallographic data for III, I and II. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce00121h |
| ‡ Crystal data: I: C38H31Cr1N1Na1O13S24, M = 1554.07, black sheet, a = 5.3536(5), b = 10.1811(9), c = 17.2836(15) Å, α = 95.641(4), β = 97.596(5), γ = 90.246(6)°, U = 929.12(14) Å3, T = 120(2) K, space group P1, Z = 2/3, μ = 1.172 mm−1, reflections collected = 4182, independent reflections = 4182, flack parameter = −0.4(2), R1 = 0.1145, wR2 = 0.2357 [F2 > 2σ(F2)], R1 = 0.2085, wR2 = 0.2806 (all data). Crystal data: II: C38H27Cr1N1Na1O12S24, M = 1534.04, green plate, a = 10.4088(4), b = 14.7524(8), c = 18.2275(8) Å, α = 90, β = 94.741(3), γ = 90°, U = 2789.3(2) Å3, T = 120(2) K, space group P21, Z = 2, μ = 1.171 mm−1, reflections collected = 21 Crystal data: III: C38H30Cr1Na1O13S24, M = 1539.05, black plate, a = 8.8372(2), b = 9.5138(2), c = 18.9128(4) Å, α = 81.9380(10), β = 77.4830(10), γ = 68.0910(10)°, U = 1382.39(5) Å3, T = 120(2) K, space group P1, Z = 1, μ = 1.182 mm−1, reflections collected = 30 All crystals were run on Bruker-Nonius KappaCCD diffractometer with Mo rotating anode, using standard control and processing software. All structures were solved and refined with programs from the SHELX family. |
| This journal is © The Royal Society of Chemistry 2015 |