
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An in situ EDXRD kinetic and mechanistic study of the hydrothermal crystallization of TiO2 nanoparticles from nitric acid peptized sol–gel
Mohammad
Rehan
ab,
Girish M.
Kale
c and
Xiaojun
Lai
*a
aInstitute of Particle Science and Engineering (IPSE), School of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK. E-mail: x.lai@leeds.ac.uk; Fax: +44 (0)113 343 2405; Tel: +44 (0)113 343 2439
bCenter of Excellence in Environmental Studies (CEES), King Abdulaziz University (KAU), Jeddah, Saudi Arabia. E-mail: dr.mohammad_rehan@yahoo.co.uk; Tel: +966 583047435
cInstitute for Materials Research (IMR), School of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK. E-mail: g.m.kale@leeds.ac.uk; Fax: +44 (0)3 343 2384; Tel: +44 (0)113 343 2805
First published on 14th January 2015
Abstract
In situ energy dispersive X-ray diffraction (EDXRD) has been used to monitor the crystallization of TiO2 nanoparticles from hydrothermal reactions of nitric acid (HNO3) peptized sol–gels at 210, 230, 250 and 270 °C. The EDXRD peaks revealed the formation of pure rutile phase TiO2 particles from all the hydrothermal reactions studied. The reaction kinetic data have been determined by monitoring changes in the integrated peak areas of the highest intensity diffraction peak (110). A kinetic and mechanistic analysis of the hydrothermal synthesis of TiO2 nanoparticles has been proposed based on the Avrami–Erofe'ev kinetic model. The Avrami exponent values obtained were within the range of around 0.5–1 for all the reactions studied, indicating that the hydrothermal crystallization of TiO2 nanoparticles involves a diffusion-controlled process mechanism. The as-synthesized TiO2 nanoparticles have been further characterized by powder X-ray diffraction (XRD) and transmission electron microscopy (TEM). The XRD data are in good agreement with the EDXRD results and confirm the formation of pure rutile phase TiO2 particles. The TEM results showed that the TiO2 particles produced are below 100 nm in size and exhibit bi-modal size and morphology distribution.
Introduction
TiO2 has attracted wide scientific and technological interest as a result of its applications in photocatalysis, optical devices, gas sensors, solar cells, and antibacterial activity, in the recent past.1–6 The possibilities of new and enhanced current applications further increase when the material is made in the nanometer size range due to its unique physical, chemical, mechanical and optical properties. This has led to many breakthroughs in the preparation, modification and applications of TiO2 nanomaterials to date.7–14The hydrothermal process, in which chemical reactions occur in aqueous or organo-aqueous media, under the simultaneous application of heat and pressure, has been widely used to produce TiO2 nanoparticles.15–23 This process has many advantages such as it uses relatively low reaction temperatures to obtain particles with narrow particle size distribution, reduces agglomeration and has good phase homogeneity and controlled particle morphology. Despite all the advantages, one major challenge of the hydrothermal process is the difficulty of direct observation of the crystals as they grow due to the black box nature of the process.24 The use of pressure vessels along with a large number of reaction variables present, including reaction temperature, time, solvent and reactant concentrations and percent fill, contributes to the lack of control in these reactions.25
In order to achieve good control of the hydrothermal synthesis of nanoparticles with precisely tailored properties, a better understanding of the reaction kinetics and growth mechanism is essential. An in situ monitoring of hydrothermal reactions using synchrotron radiation EDXRD has been used to obtain information to probe the reaction kinetics and mechanisms.26,27 The high photon energy white X-ray beam of synchrotron radiation has the ability to penetrate into the stainless steel pressure vessel, providing time-resolved diffraction patterns of crystalline materials on an energy scale. This allows an entire diffraction pattern of the contents of the reaction vessel to be collected in situ in a short time span (e.g. 60 s).28 The non-invasive EDXRD technique has been developed as a powerful tool at Synchrotron Radiation Source (SRS) station 16.4 at Daresbury Laboratory with various applications of in situ process observations such as process tomography, hydrothermal crystallization microporous materials synthesis, chemical reactions, ambient pressure crystallization, and crystal phase transformation reactions.29–40 In addition to these, some other applications of in situ techniques to study the sol–gel synthesis and crystallization reactions have also been reported recently.41–49
In this paper, we present the use of in situ EDXRD at station 16.4 of Daresbury Laboratory SRS to study the hydrothermal synthesis of TiO2 nanoparticles. The nanoparticles have been synthesized by hydrothermal reactions of HNO3 peptized sol–gels at 210, 230, 250 and 270 °C. The as-synthesized nanoparticles have also been characterized by XRD and TEM analyses. A kinetic and mechanistic analysis of the hydrothermal synthesis of TiO2 nanoparticles based on the Avrami–Erofe'ev kinetic model has also been reported.
Experimental
Titanium butoxide (Ti(OBu)4) used as a Ti precursor, 2-propanol ((CH3)2CHOH) used as solvent and nitric acid (HNO3, 70%) used as a peptizing agent were all obtained from Sigma-Aldrich, UK and used without further purification. A 0.5 M solution of titanium butoxide in 2-propanol was prepared and then added dropwise to distilled water at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 volume ratio with continuous stirring for about 1 hour. The obtained suspension was filtered to collect the white precipitate of hydrolysed TiO2. The HNO3 peptized sol–gel was then prepared by stirring a mixture of 31 g of the TiO2 precipitate, 2.7 ml of HNO3 (70%) and 90 g of distilled water for about 45 min. This produced a clear pale yellow coloured sol–gel. These sol–gels were then treated hydrothermally at different temperatures to produce TiO2 nanoparticles.
:4 volume ratio with continuous stirring for about 1 hour. The obtained suspension was filtered to collect the white precipitate of hydrolysed TiO2. The HNO3 peptized sol–gel was then prepared by stirring a mixture of 31 g of the TiO2 precipitate, 2.7 ml of HNO3 (70%) and 90 g of distilled water for about 45 min. This produced a clear pale yellow coloured sol–gel. These sol–gels were then treated hydrothermally at different temperatures to produce TiO2 nanoparticles.
The hydrothermal synthesis of TiO2 nanoparticles from HNO3 peptized sol–gels was monitored in situ using EDXRD in Station 16.4 of the SRS at Daresbury Laboratory, UK. The details of the station and the experimental setup can be found in our previous work.50 In a typical hydrothermal process, 20 ml of the HNO3 peptized sol–gel was placed in a 30 ml PTFE lined pressure vessel and heated to desired temperature. A special inconel thinner-walled pressure vessel was used to minimize the absorption of X-rays. The hydrothermal reaction mixture was stirred continuously using a PTFE coated magnetic stirrer placed in the vessel to ensure that the particles produced remained in the pathway of the beam throughout the reaction and did not settle down. Four hydrothermal reactions were studied at four different temperatures of 210, 230, 250 and 270 °C with a heating rate of ~10 °C min−1.
During each hydrothermal reaction, the X-ray diffraction patterns were simultaneously collected every 60 s on an energy scale using three-element solid-state detectors; top, middle and bottom, fixed at 2θ angles of 7.375°, 4.51° and 1.61°, respectively. The three-element solid state detector setup used in this work is described elsewhere.51,52 The raw data recorded by Pincer software during the course of hydrothermal reactions were in channel numbers, which were converted to the energy scale (keV) using a program called DL Convertor. The patterns on the energy scale were then converted to the d-spacing scale (Å) using the modified form of Bragg's law:
| E = 6.199/(d sinθ) | (1) |
All hydrothermal reaction products were further characterized using powder XRD and TEM. The equipment, sample preparation and conditions used for this characterization were exactly the same as those described in our previous work.50
Results and discussion
The in situ EDXRD spectra collected during the hydrothermal synthesis of TiO2 nanoparticles from HNO3 peptized sol–gels treated at 210, 230, 250 and 270 °C are shown in Fig. 1(a)–(d) respectively. As the hydrothermal reactions proceeded, the pressure of each vessel started to build up after 30 to 40 min and then stabilized after the attainment of the set temperature. The maximum pressure values observed were around 20, 34, 40 and 42 bar for reactions at 210, 230, 250 and 270 °C, respectively. | ||
| Fig. 1 In situ EDXRD data collected during the hydrothermal synthesis of TiO2 nanoparticles from HNO3 peptized sol–gels at (a) 210 °C, (b) 230 °C, (c) 250 °C and (d) 270 °C. Modified versions of our previously published EDXRD graphs.50 | ||
In all experiments, the first few EDXRD spectra only showed a broad hump between 1.5 to 4.5 Å d-spacing with no Bragg peaks. This hump is produced by the diffuse scattering of the complex structured gel in the reaction vessel and the spectral intensity profile of the white beam.29 During the hydrothermal reaction of HNO3 peptized sol–gel at 210 °C, the first diffraction peak emerged with a d-spacing value of 3.26 Å after an induction time of 34 min, corresponding to the (110) plane of the rutile crystal structure (Fig. 1a). As the reaction proceeded further, the intensity of this peak increased gradually until around 90 min and then almost stabilized for the rest of the reaction time. Another peak with lower intensity was noticed shortly after the first one with a d-spacing value of 2.50 Å, corresponding to the (101) plane of the rutile crystal structure. The intensity of this second peak (101) remained lower than the first one (110), throughout the whole reaction time.
The hydrothermal processes heated up to 230, 250 and 270 °C also showed very similar peak emergence and development akin to that observed at 210 °C. The two same Bragg peaks of d-spacings 3.26 and 2.50 Å, corresponding to rutile (110) and (101) diffraction planes, respectively, were observed. However the induction times, 33, 31 and 30 min for the processes at 230, 250 and 270 °C, respectively, were slightly lower than that for the process at 210 °C. Furthermore, the overall peak intensities for the processes at higher temperatures are greater than the process at the lower temperature, which can be attributed to the larger number and size of particles produced at higher process temperature.
There were two small diffraction peaks noticed at d-spacings 2.10 and 2.19 Å for the whole reaction period of 250 and 270 °C (Fig. 1(c) and (d)), which do not match with any of the rutile diffraction planes in the reference patterns. These peaks are probably arising from the fluorescence or escape peaks associated with synchrotron beam conditions on the day of the experiment. The data shown in Fig. 1(a) and (b) were obtained on a different experimental day and hence these two additional peaks are not present. The EDXRD patterns show that the rutile phase TiO2 particles are directly produced from the sol–gel complex structure upon hydrothermal treatment without formation of any crystalline intermediate phase.
The extent of reaction (α) has been measured using the integrated areas of the highest intensity peak (110) and plotted against the reaction time for each hydrothermal process studied (Fig. 2a–d). The peak area is integrated using the Gaussian function by an in-house developed program (XRD Peak Area Analyser). These values can be converted to extent of reaction (α), scaled from zero to one, using the relationship:
| α(t) = Ihkl(t)/Ihkl(max) | (2) |
 | ||
| Fig. 2 Extent of reaction against time for the (110) peak of TiO2 particles produced by in situ hydrothermal processes at 210, 230, 250 and 270 °C (a–d) and their corresponding Sharp and Hancock plots showing the kinetic data (e–h), respectively. | ||
The quantitative kinetic analysis of the hydrothermal synthesis of TiO2 nanoparticles produced from HNO3 peptized sol–gels was achieved by using the nucleation–growth kinetic model described by Avrami–Erofe'ev.53–55 This model is widely used for phase transitions and crystal growth processes in chemistry (eqn (3))
| α = 1–exp[−(k(t − t0))m] | (3) |
| ln[−ln(1 − α)] = mln(t − t0) + mln(k) | (4) |
Thus a plot of ln[–ln(1 − α)] against ln(t − t0) should yield a straight line with a gradient m and an intercept mln(k), if the Avrami–Erofe'ev model is valid. The Sharp–Hancock plots based on the kinetic data derived from the (110) reflection peaks of TiO2 (Fig. 2a–d) synthesized by in situ hydrothermal processes at 210, 230, 250 and 270 °C are shown in Fig. 2(e–h), respectively. The resultant plots exhibit a linear relationship within the range of 0.2 < α < 0.8, and the gradient (m) and intercepts (mln(k)) of lines have been determined by linear regression to compute the kinetic parameters (Table 1). The linear regression analysis gives an average error bar for each data point and the overall average error decreases as the reaction temperature increases from 210 °C to 270 °C.
| T (°C) | t 0 (s) | k (s−1) | m |
|---|---|---|---|
| 210 | 2020 | 3.95 × 10−4 | 0.53 |
| 230 | 1950 | 6.03 × 10−4 | 0.44 |
| 250 | 1880 | 4.14 × 10−4 | 0.69 |
| 270 | 1810 | 2.55 × 10−4 | 0.76 |
It can be seen in Table 1 that the calculated values of m are 0.53, 0.44, 0.69 and 0.76 for the hydrothermal process of TiO2 at 210, 230, 250 and 270 °C, respectively. Since the values of m are roughly within 0.5–1, it indicates that the hydrothermal reaction for the formation of rutile TiO2 from the precursor gel occurs via a diffusion-controlled mechanism.56 This means that the rate of diffusion of the reactants through the solution to the site of crystallization is the rate limiting factor of the overall reaction. The rates of all the other steps including breaking of the sol–gel structure upon heating to release the reactive species, formation of nuclei and crystal growth are relatively faster and therefore do not affect the overall rate of the reaction for the formation of TiO2 nanoparticles under hydrothermal conditions.
It should be noted that the kinetic parameters k and m calculated from the Avrami–Erofe'ev kinetic model are reasonably consistent and within appropriate ranges, even though several factors may have influenced their accuracy during the experimental and data analysis stages. This includes, but not limited to, the complex nature of sol–gel used, EDXRD data collection over few visits to SRS station 16.4, possible variations in stirring speed of different cells used, detection of small diffraction peaks and errors in induction time due to the lower detection limits intrinsic to the EDXRD technique,28 and errors in integration of the peak area which is a key parameter used for calculating the extent of reaction and other kinetic and mechanistic parameters. It is also worth mentioning here that the cooling process after the completion of the hydrothermal reactions can also be an important phenomenon. In this study, natural air cooling, which takes few hours, has been adapted for all the hydrothermal processes, but this cooling process was not monitored in situ due to limited availability of beamtime. The authors believe that natural air cooling has very little effect, if any, on crystal formation and growth processes. However, it would still be interesting to monitor in situ any changes during the cooling process of hydrothermal systems.
The products obtained from the in situ hydrothermal reactions carried out at Daresbury Laboratory have been further characterized by powder XRD and TEM to study the crystal structure, size and morphology. The XRD patterns of the oven dried powder samples, from in situ hydrothermal reactions carried out at Daresbury Laboratory at 210 °C, 230 °C and 270 °C, are shown in Fig. 3. This confirms that rutile phase TiO2 has been produced from all the hydrothermal processes studied. The XRD peaks are indexed using the reference pattern JCPDS card 01-088-1172. There is no evidence of any other crystalline phase or impurity in any of the samples. These powder XRD results are in good agreement with our in situ EDXRD results. The XRD pattern for the sample produced at 250 °C is missing because this sample was misplaced during laboratory shift and the repeat experiment was not possible due to unavailability of any further beamtime for this project. However, the authors have absolutely no doubt on this sample forming the same rutile phase TiO2 particles because many ex situ hydrothermal synthesis experiments under similar process conditions have been conducted at the University of Leeds laboratories and the results are in complete agreement.
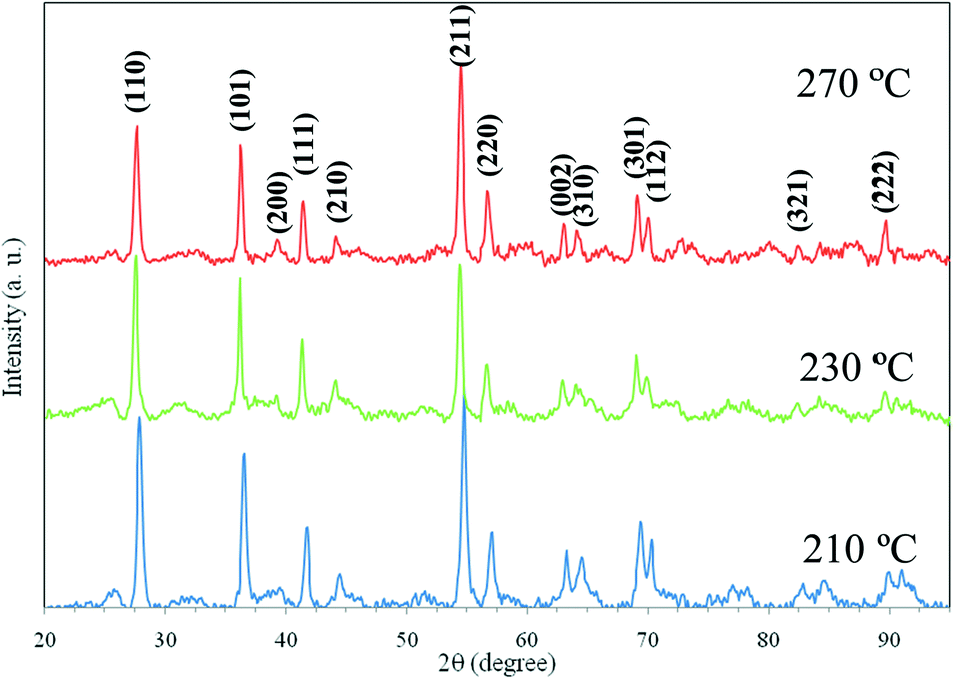 | ||
| Fig. 3 XRD patterns of TiO2 nanoparticles produced by in situ hydrothermal reactions of HNO3 peptized sol–gels at 210 °C, 230 °C and 270 °C. Modified versions of our previously published XRD patterns.50 | ||
The TEM images of the TiO2 nanoparticles produced by in situ hydrothermal reactions of HNO3 peptized sol–gels at 210, 230, 250 and 270 °C are shown in Fig. 4a–d, respectively. It is evident from these images that the particles produced from all these processes generally exhibit bi-modal size and morphological features. The smaller spherical particles are found to be in the average size range of around 5–15 nm and the larger rod-like particles around 30–95 nm. The particles produced from the process at the highest temperature of 270 °C showed relatively narrower particle size and morphology distributions with fewer smaller spherical particles compared with the other processes studied at the lower temperatures.
 | ||
| Fig. 4 TEM images of TiO2 nanoparticles produced by in situ hydrothermal reactions of HNO3 peptized sol–gels at (a) 210 °C, (b) 230 °C, (c) 250 °C and (d) 270 °C. | ||
The in situ EDXRD technology coupled with synchrotron radiation provides the capability to study reactions in very special conditions such as under very low or high temperatures and/or pressures, allowing very fast and time resolved measurements. This makes it possible to carry out detailed kinetic and mechanistic studies, which are not possible with conventional XRD. However, there are some limitations to this technology, for example it relies on the ability of the high energy beam of synchrotron radiation to penetrate through the steel walls of the pressure vessel. The thickness of the pressure vessel to be used, which controls the maximum safe working temperature and pressure conditions, depends on the maximum usable energy of synchrotron radiation, because the higher the X-ray energy the more penetrating the beam.58 So far we have studied the hydrothermal reactions at a maximum temperature of 270 °C and pressure of 42 bar using a pressure vessel with a wall thickness of 0.25 mm at station 16.4 Daresbury Laboratory. However, different beamlines throughout the world have different levels of energies and capabilities that determine the maximum safe working temperature and pressure conditions at which one can study any hydrothermal system. Furthermore, in situ EDXRD is less sensitive due to high noise from the scattering background and has lower resolution than the conventional angular dispersive XRD. This is a general issue of in situ EDXRD techniques, which also makes particle sizing unfeasible.
Conclusions
In situ EDXRD has been successfully used to study the evolution of crystalline TiO2 nanoparticles from HNO3 peptized sol–gels at 210, 230, 250 and 270 °C in the hydrothermal process. Pure rutile TiO2 nanoparticles have been produced as confirmed by both in situ EDXRD and powder XRD data. The reaction kinetics and mechanistic analysis of the hydrothermal synthesis of TiO2 nanoparticles has been proposed based on the Avrami–Erofe'ev nucleation–growth kinetic model. It has been found that the hydrothermal synthesis of TiO2 nanoparticles from the HNO3 peptized sol–gel process exhibits a diffusion-controlled reaction mechanism. The TEM results have shown that the particles produced are below 100 nm in size and exhibit bi-modal particle size and morphology distribution. The fundamental understanding of reaction kinetics and the mechanism developed from these studies are very important for controlled synthesis of tailor-made nanoparticles employing the hydrothermal process.Acknowledgements
The authors are thankful to EPSRC for funding the project, CCLRC for allowing access to SRS station 16.4 (Daresbury Laboratory, UK), IPSE and IMR (University of Leeds) for providing facilities and infrastructure to carry out this research investigation. The authors also thank D. J. Taylor and T. M. Bell, station scientists in station 16.4, for their assistance in setting up and running the synchrotron radiation experiments, and M. Ward for his assistance with the TEM analysis.References
- R. Wang, K. Hashimoto, A. Fujishima, M. Chikuni, E. Kojima, A. Kitamura, M. Shimohigoshi and T. Watanabe, Nature, 1997, 388, 431 CrossRef CAS PubMed.
- N. O. Savage, S. A. Akbar and P. K. Dutta, Sens. Actuators, B, 2001, 72, 239 CrossRef CAS.
- Y. Shimizu, N. Kuwano, T. Hyodo and M. Egashira, Sens. Actuators, B, 2002, 83, 195 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- K. Sunada, Y. Kikuchi, K. Hashimoto and A. Fujishima, Environ. Sci. Technol., 1998, 32, 726 CrossRef CAS.
- A. Fujishima, T. N. Rao and D. A. Tryk, Electrochim. Acta, 2000, 45, 4683 CrossRef CAS.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1 CrossRef CAS.
- D. A. Tryk, A. Fujishima and K. Honda, Electrochim. Acta, 2000, 45, 2363 CrossRef CAS.
- A. Hagfeldt and M. Graetzel, Chem. Rev., 1995, 95, 49 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Y. Jr, Chem. Rev., 1995, 95, 735 CrossRef CAS.
- A. Millis and S. L. Hunte, J. Photochem. Photobiol., A, 1997, 108, 1 CrossRef.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed.
- Y. Oguri, R. E. Riman and H. K. Bowen, J. Mater. Sci., 1998, 23, 2897 CrossRef.
- R. R. Bacsa and M. Grätzel, J. Am. Ceram. Soc., 1996, 79, 2185 CrossRef CAS PubMed.
- H. Cheng, J. Ma, Z. Zhao and L. Qi, Chem. Mater., 1995, 7, 663 CrossRef CAS.
- J. Yang, S. Mei and J. M. F. Ferreira, J. Am. Ceram. Soc., 2000, 83, 1361 CrossRef CAS PubMed.
- J. Yang, S. Mei and J. M. F. Ferreira, J. Am. Ceram. Soc., 2001, 84, 1696 CrossRef CAS PubMed.
- J. Yang, S. Mei and J. M. F. Ferreira, J. Mater. Res., 2002, 17, 2197 CrossRef CAS.
- J. Yang, S. Mei, J. M. F. Ferreira, P. Norby and S. Quaresma, J. Colloid Interface Sci., 2005, 283, 102 CrossRef CAS PubMed.
- K. Yanagisawa, Y. Yamamoto, Q. Feng and N. Yamasaki, J. Mater. Res., 1998, 13, 825 CrossRef CAS.
- H. Lee and G. M. Kale, Int. J. Appl. Ceram. Technol., 2008, 5, 657 CrossRef CAS PubMed.
- K. Byrappa and M. Yoshimura, Handbook of Hydrothermal Technology, ed. W. Andrew, Elsevier, Oxford and Waltham, 2nd edn, 2013, ch. 1, pp. 34–36 Search PubMed.
- A. J. Norquist and D. O'Hare, J. Am. Chem. Soc., 2004, 126, 6673 CrossRef CAS PubMed.
- A. K. Cheetham and C. F. Mellot, Chem. Mater., 1997, 8, 2269 CrossRef.
- R. J. Francis and D. O'Hare, J. Chem. Soc., Dalton Trans., 1998, 3133 RSC.
- N. Yee, S. Shaw, L. G. Benning and T. H. Nguyen, Am. Mineral., 2006, 91, 92 CrossRef CAS.
- S. M. Clark, Nucl. Instrum. Methods Phys. Res., Sect. A, 1996, 381, 161 CrossRef CAS.
- S. D. M. Jacques, K. Pile, P. Barnes, X. Lai, K. J. Roberts and R. A. Williams, Cryst. Growth Des., 2005, 5, 395 CAS.
- R. J. Francis, S. J. Price, J. S. O. Evans, S. O'Brien and D. O'Hare, Chem. Mater., 1996, 8, 2102 CrossRef CAS.
- D. O'Hare, J. S. O. Evans, R. J. Francis, P. S. Halasyamani, P. Norby and J. Hanson, Microporous Mesoporous Mater., 1998, 21, 253 CrossRef.
- F. Millange, R. I. Walton, N. Guillou, T. Loiseau, D. O'Hare and G. Ferey, Chem. Mater., 2002, 14, 4448 CrossRef CAS.
- S. Shaw, C. Michael, B. Henderson and S. M. Clark, Am. Mineral., 2002, 87, 533 CAS.
- C. L. Cahill, L. G. Benning, H. L. Barnes and J. B. Parise, Chem. Geol., 2000, 167, 53 CrossRef CAS.
- J. S. O. Evans, S. J. Price, H. V. Wong and D. O'Hare, J. Am. Chem. Soc., 1998, 120, 10837 CrossRef CAS.
- A. M. Fogg, J. S. Dunn and D. O'Hare, Chem. Mater., 1998, 10, 356 CrossRef CAS.
- A. M. Fogg, J. S. Dunn, S. G. Shyu, D. R. Cary and D. O'Hare, Chem. Mater., 1998, 10, 351 CrossRef CAS.
- R. J. Francis, S. J. Price, S. Obrien, A. M. Fogg, D. O'Hare, T. Loiseau and G. Ferey, Chem. Commun., 1997, 521 RSC.
- S. Obrien, R. J. Francis, S. J. Price, D. O'Hare, S. M. Clark, N. Okazaki and K. Kuroda, J. Chem. Soc., Chem. Commun., 1995, 2423 RSC.
- K. A. Cheetham and F. M. Caroline, Chem. Mater., 1997, 9, 2269 CrossRef.
- N. Blagden, R. Davey, M. Song, M. Quayle, S. Clark, D. Taylor and A. Nield, Cryst. Growth Des., 2003, 3, 197 CAS.
- A. Jianu, L. Stanciu, J. R. Groza, C. Lathe and E. Burkel, Nucl. Instrum. Methods Phys. Res., Sect. B, 2003, 199, 44 CrossRef CAS.
- L. B. Kirsch, E. K. Richman, A. E. Riley and S. H. Tolbert, J. Phys. Chem. B, 2004, 108, 12698 CrossRef.
- D. R. Hummer, J. D. Kubicki, P. R. C. Kent, J. E. Post and P. J. Heaney, J. Phys. Chem. C, 2009, 113, 4240 CAS.
- R. J. Francis and D. O'Hare, J. Chem. Soc., Dalton Trans., 1998, 3133 RSC.
- Y. Zhou, E. Antonova, W. Bensch and G. R. Patzke, Nanoscale, 2010, 2, 2412 RSC.
- Y. Zhou, E. Antonova, W. Bensch and G. R. Patzke, Nanoscale, 2010, 2, 2412 RSC.
- Y. Zhou, E. Antonova, Y. Lin, J. D. Grunwaldt, W. Bensch and G. R. Patzke, Eur. J. Inorg. Chem., 2012, 783 CrossRef CAS.
- M. Rehan, X. Lai and G. M. Kale, CrystEngComm, 2011, 13, 3725 RSC.
- P. Barnes, A. C. Jupe, S. L. Colston, S. D. Jacques, A. Grant, T. Rathbone, M. Miller, S. M. Clark and R. J. Cernik, Nucl. Instrum. Methods Phys. Res., Sect. B, 1998, 134, 310 CrossRef CAS.
- S. L. Colston, S. D. M. Jacques, P. Barnes, A. C. Jupe and C. Hall, J. Synchrotron Radiat., 1998, 5, 112 CrossRef CAS PubMed.
- M. Avrami, J. Chem. Phys., 1939, 7, 1103 CrossRef CAS PubMed.
- M. Avrami, J. Chem. Phys., 1940, 8, 212 CrossRef CAS PubMed.
- M. Avrami, J. Chem. Phys., 1941, 9, 177 CrossRef CAS PubMed.
- S. F. Hulbert, J. Br. Ceram. Soc., 1969, 6, 11 CAS.
- J. D. Hancock and J. H. Sharp, J. Am. Ceram. Soc., 1972, 55, 74 CrossRef CAS PubMed.
- S. Shaw, C. M. B. Henderson, S. M. Clark, Y. Wang, The Proceedings of Goldschmidt Conference, Toulouse, France, 1998, pp. 1377 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |