
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structure determination, and formation of a theobromine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :oxalic acid 2:1 cocrystal†
:oxalic acid 2:1 cocrystal†
Franziska
Fischer
ab,
Gudrun
Scholz
b,
Lisa
Batzdorf
ab,
Manuel
Wilke
ab and
Franziska
Emmerling
*a
aBAM Federal Institute for Materials Research and Testing, Richard-Willstaetter-Str. 11, 12489 Berlin, Germany. E-mail: franziska.emmerling@bam.de
bDepartment of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany
First published on 19th November 2014
Abstract
The structure and the formation pathway of a new theobromine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oxalic acid (2:1) cocrystal are presented. The cocrystal was synthesised mechanochemically and its structure was solved based on the powder X-ray data. The mechanochemical synthesis of this model compound was studied in situ using synchrotron XRD. Based on the XRD data details of the formation mechanism were obtained. The formation can be described as a self-accelerated (‘liquid like’) process from a highly activated species.
:oxalic acid (2:1) cocrystal are presented. The cocrystal was synthesised mechanochemically and its structure was solved based on the powder X-ray data. The mechanochemical synthesis of this model compound was studied in situ using synchrotron XRD. Based on the XRD data details of the formation mechanism were obtained. The formation can be described as a self-accelerated (‘liquid like’) process from a highly activated species.
Introduction
The properties of active pharmaceutical ingredients (APIs) are typically not optimised for their planned applications.1–3 This is one of the crucial issues in the development of new pharmaceuticals. Many APIs show insufficient bioavailability, which is closely related to their low water solubility. In addition, the polymorphism of drugs has to be considered. Different modification of a given compound can be formed due to similar enthalpies of formation. The crystal structure of a material has a determining influence on its physicochemical properties including melting point, stability against physical and chemical stress, dissolution behaviour, solubility, and bioavailability.4Polymorphs of an API can show changes in the properties or even in the therapeutic effects.5–7 Therefore, it remains a key challenge to improve the physicochemical properties of a drug. Crystal engineering considerations provide a possibility to overcome this issue.8 The formation of salts or solvates of APIs is a typical approach to circumvent the problem of low solubility.9–11 In this context, cocrystals of a given API have gained considerable interest in recent years. Cocrystals are crystalline phases with two or more components consisting of uncharged organic compounds, which interact via intermolecular forces.2,12–16 As a result of cocrystallisation, new crystal structures with new physicochemical properties compared to those of the API emerge.17–21
Cocrystals can be synthesised by different methods; typically solution based techniques are used. However, these methods present some disadvantages. For example, the solution based cocrystal formation requires a comparable solubility of the educts for successful synthesis. Due to the poor solubility of the APIs large amounts of solvent are needed. Additionally, solvent molecules could be incorporated in the crystalline structure of the cocrystal, which complicates the control of the product.3,22
Mechanochemistry is an elegant way to circumvent these problems. Typically no or only small amounts of solvent are needed for the milling reactions. Furthermore, the reactions are very fast, nearly quantitative, and proceed without the formation of by-products. Several cocrystals which are not accessible via conventional methods could only be synthesised mechanochemically.23–26 Consequently, mechanochemical syntheses have been used increasingly in the past years. A detailed understanding of the underlying mechanism of the mechanochemical syntheses is still scarce.27,28 Recently, Friščić et al. introduced real-time and in situ monitoring of milling reactions using a mixer mill. These experiments provided the first direct insight into the formation pathways.29–31 Here, we present the first in situ XRD study of milling synthesis using a ball mill setup.
As a model system, a new 2:1 cocrystal of the API theobromine (tb) and the coformer oxalic acid (ox) was used in the milling experiments. The in situ investigation of the reaction was conducted in a Perspex grinding jar using synchrotron XRD. Based on the obtained data, a diffusion mechanism was postulated.
Results and discussion
The tb:ox cocrystal in a molar ratio of 2:1 was synthesised by neat grinding. In contrast to the structurally similar APIs theophylline and caffeine only a few theobromine cocrystals are known.3,32,33 The theobromine:oxalic acid (tb:ox) cocrystal represents an interesting model compound for further investigations of the formation pathway during the mechanochemical syntheses. The powder X-ray diffraction (PXRD) pattern of the new compound is depicted in Fig. 1a in comparison to the PXRD patterns of the reactants tb and ox dihydrate. The powder pattern of the cocrystal shows no contributions from the reflections of the reagents, indicating a complete reaction.
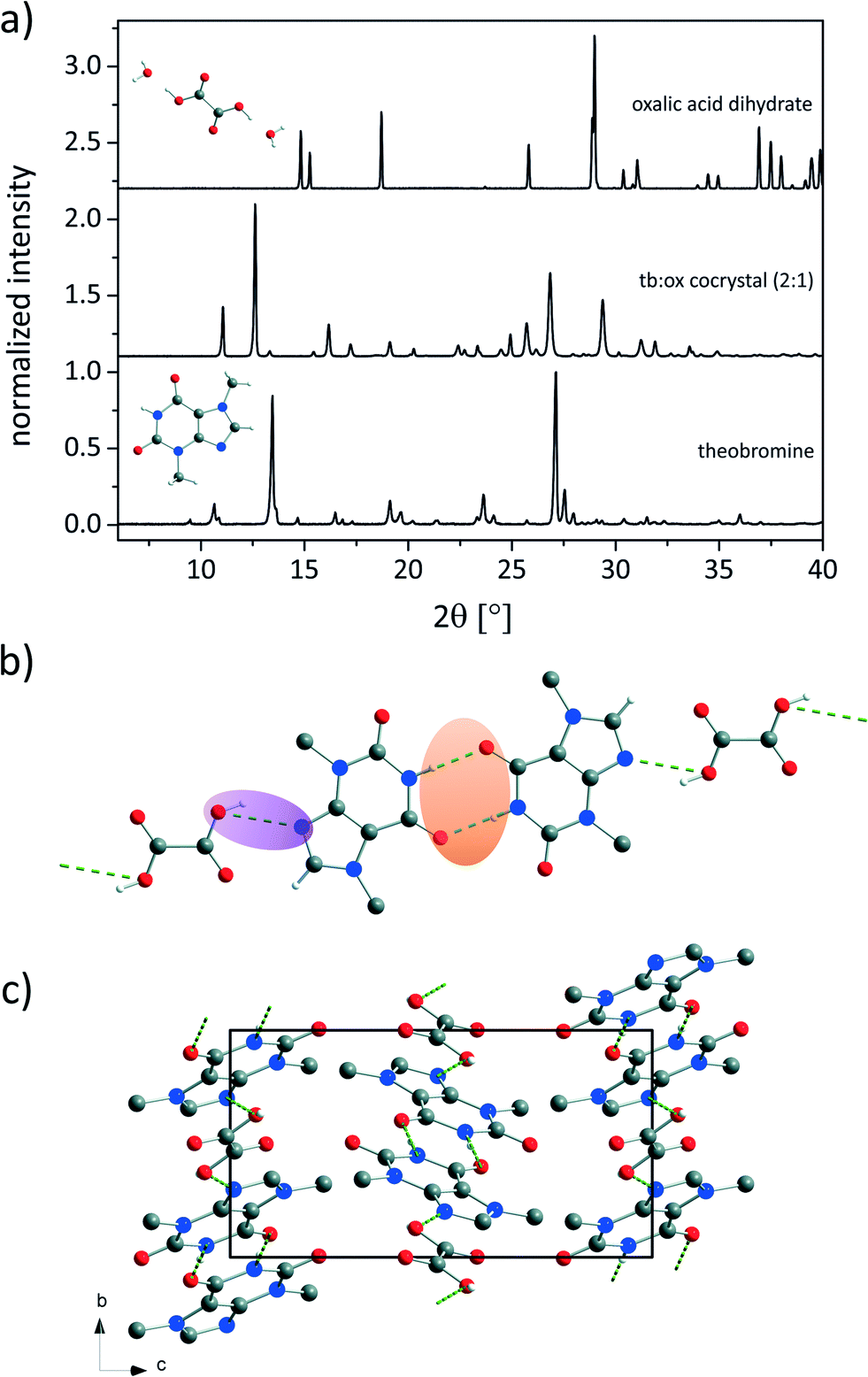 | ||
| Fig. 1 a) Powder XRD patterns of the tb:ox cocrystal (center) and the reactants theobromine (bottom) and oxalic acid dihydrate (top). The background contributions of the sample holders were corrected. b) Bonding arrangement and c) structure of the 2:1 cocrystal tb:ox seen along the a-axis. The hydrogen atoms not involved in the hydrogen bonding were omitted for clarity. Green dashed lines indicate hydrogen bonds. b) The structure of the 2:1 cocrystal tb:ox seen along the a-axis. The hydrogen atoms not involved in the hydrogen bonding were omitted for clarity. Green dashed lines indicate hydrogen bonds. | ||
Based on the powder pattern determination of the cocrystal structure followed by Rietveld refinement was possible. The resulting structure is presented in Fig. 1b. The corresponding Rietveld refinement is shown in Fig. 2 indicating good agreement between the simulated and measured powder patterns. The tb:ox cocrystal crystallises in the monoclinic space group P21/c (a = 8.89209(45) Å, b = 7.50930(28) Å, c = 15.60777(84) Å, β = 116.5691(38)°, V = 932.124(83) Å3). Each tb molecule is connected to a tb molecule and an ox molecule via hydrogen bonds. One hydrogen bond is formed between the nitrogen atom of the secondary amine of a tb molecule and the oxygen atom from a carbonyl group of another tb molecule (N–H⋯O, dH⋯A = 2.803 Å, dD⋯A = 1.92 Å, ∠D–H⋯A = 170°), resulting in an R22(8) dimer (orange). An additional hydrogen bond (violet) is formed between the tertiary amine of a tb molecule and the hydroxyl group of an ox carboxyl group (O–H⋯N, dH⋯A = 2.843 Å, dD⋯A = 2.12 Å, ∠D–H⋯A = 136°), leading to a twisted chain motif running along the b-axis.
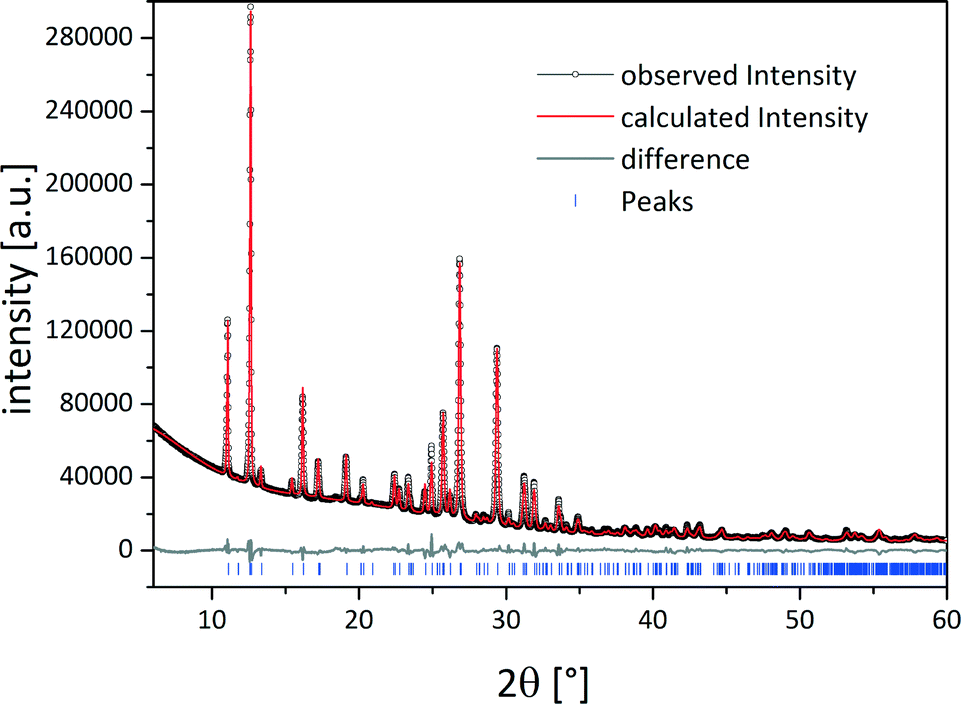 | ||
| Fig. 2 Rietveld refinement of the crystal structure of the tb:ox cocrystal. Scattered X-ray intensity for the tb:ox 2:1 cocrystal under ambient conditions as a function of the diffraction angle 2θ. The observed pattern (black circles), the best Rietveld fit profile (red line), the reflection positions (blue tick marks), and the difference curve (grey line) between the observed and calculated profiles are shown. The wavelength was λ = 1.54056 Å (Cu Kα1). The R-values are Rp = 3.2% and Rwp = 4.9%; Rp, and Rwp refer to the Rietveld criteria of fit for profile and weighted profile defined by Langford and Louer.34 | ||
The absence of water in the crystal structure is evident from the DTA-TGA measurements (Fig. S3†). The first DTA signal of the cocrystal arises at a temperature of 252 °C. At this temperature the ox molecules decompose. Since the decomposition temperature of ox is 50 K higher than that of the pure sample, it can be concluded that the ox molecules are stabilised in the cocrystal.
Based on PXRD data, the position of the hydrogen atoms cannot be determined unambiguously. In order to exclude salt formation the cocrystal was investigated by Raman (Fig. S1†) and solid-state NMR (ssNMR) spectroscopy (Fig. S2†). In the Raman spectra only the band attributed to the carboxylate deformation vibration of ox dihydrate at 478 cm−1 shows a pronounced shift.35 Therefore protonation of the tb molecules in the cocrystal can be excluded. The strong shift of the carboxylate band indicates that the ox molecules interact more strongly with water in ox dihydrate than with the tb molecule in the cocrystal. This assumption is supported by the ssNMR measurements. The only ssNMR signal, which shifts considerably in the cocrystal, is due to the protons of the ox molecules at 17.0 ppm. The shift to 14.2 ppm suggests that the protons of ox are not as strongly bridged in the cocrystal as in pure ox dihydrate. Consequently, it can be assumed that the ox molecules are uncharged in the cocrystal. The water signal at 5.5 ppm disappears in the cocrystal, which reveals that no water molecules are incorporated in the cocrystal. Moreover, the observed line broadening in the 1H MAS NMR spectrum of the cocrystal (Fig. S2,† middle) supports the assumption of the formation of a network of additional hydrogen bonds.
The formation of the cocrystal was observed with in situ synchrotron XRD. The in situ experiments were conducted at the microfocus beamline μSpot (BESSY II, Helmholtz Centre Berlin for Materials and Energy, Germany) in transmission geometry using a wavelength of 1.0000 Å. A Perspex grinding jar (Fig. 3) was used as the reaction vessel. Powder diffraction patterns of the reaction mixture can be measured directly without opening the grinding jar.36 XRD patterns were acquired every 30 s during the milling process.
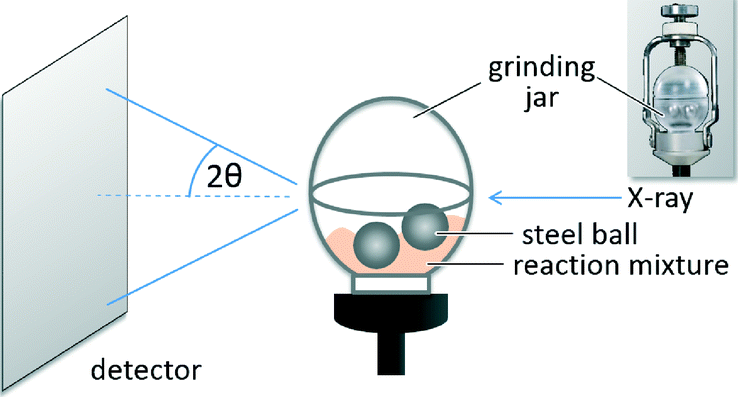 | ||
| Fig. 3 Experimental setup for the collection of the powder patterns during the neat grinding synthesis of the tb:ox cocrystal. The sketch shows a Perspex grinding jar filled with two 10 mm stainless steel grinding balls. | ||
An investigation of the mechanochemical cocrystal formation pathway was possible since the powder patterns of the cocrystal and the reactants revealed highly distinguishable, characteristic reflections. Fig. 4 shows the time resolved powder patterns obtained during the neat grinding of tb and ox dihydrate over a time span of 20 minutes. The milling reaction can be divided into three phases. In the first step only the reflections of the reactants tb and ox dihydrate are observed in the XRD patterns (phase 1). In the first 12 min the continuous, slow decomposition of the crystal structure of the reactants is traceable on the basis of the decreasing intensity of the tb reflection at 13.5° (Fig. S8†). Afterwards the fast formation of the cocrystal proceeds within 60 s (phase 2). In this second phase the reflections of the reactants are still detectable and decrease quickly with prolonged milling times. Neither the formation of a transient intermediate species nor prolonged amorphisation of the reaction mixture could be observed during this phase. The last phase begins at a milling time of 13.5 min. At this point there are no crystalline educts detectable in the reaction mixture (phase 3).
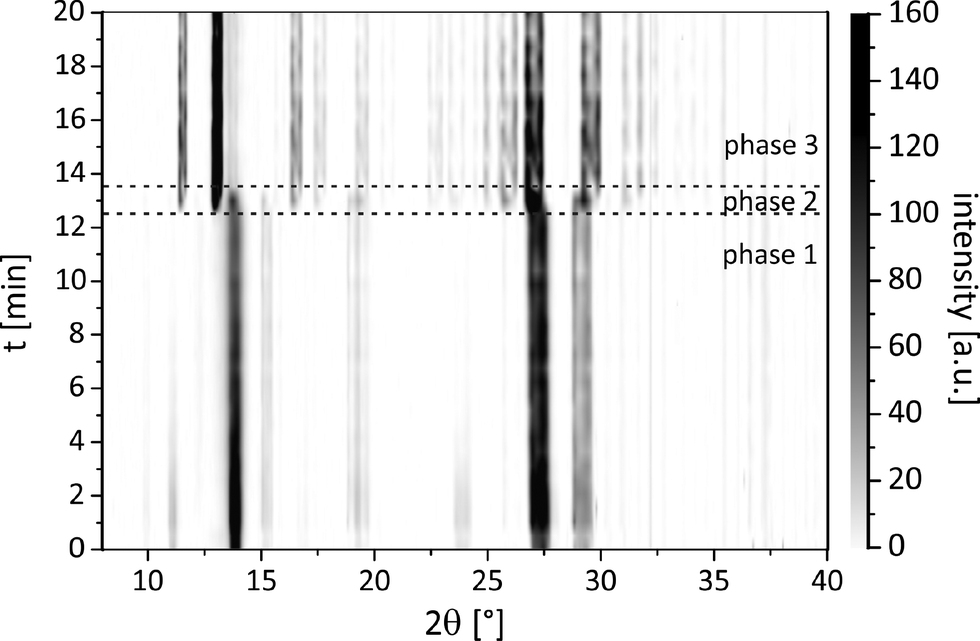 | ||
| Fig. 4 Time resolved pathway of the powder patterns during the neat grinding synthesis of the tb:ox cocrystal. | ||
Different explanations for the mechanochemical reactions are discussed in the literature and three theories have to be considered: i) the hot spot theory, ii) the magma-plasma model, and iii) reaction via diffusion. The hot spot theory is based on the assumption that the attrition between the surfaces causes local temperatures to rise above 1000 °C for short periods (10−3 to 10−4 s) on a molecular dimension.37–39 The magma-plasma model involves local temperatures about 104 °C leading to transient plasma and the ejection of energy.37,40 The third approach emphasises the importance of short diffusion pathways driven by excellent mixing of the reactants and accelerated reactions.41 No clear indication of a mechanism based on one of the three models could be found for the investigated synthesis. Keeping in mind that the diffusion coefficient in the solid state (D ≈ 10−16 m2 s−1) is significantly lower than the diffusion coefficient in fluid phases (D ≈ 10−9 m2 s−1) a comparison of the conditions during milling with a liquid-like situation appears obvious.
The fast transformation can be explained by a self-accelerated process from a highly activated species that is completed in 60 s. This process leads to the direct conversion of the solid reactants to the product. There is no driving force based on salt formation or protonation since the cocrystal consists only of neutral molecules. The derived formation pathway is illustrated in Fig. 5.
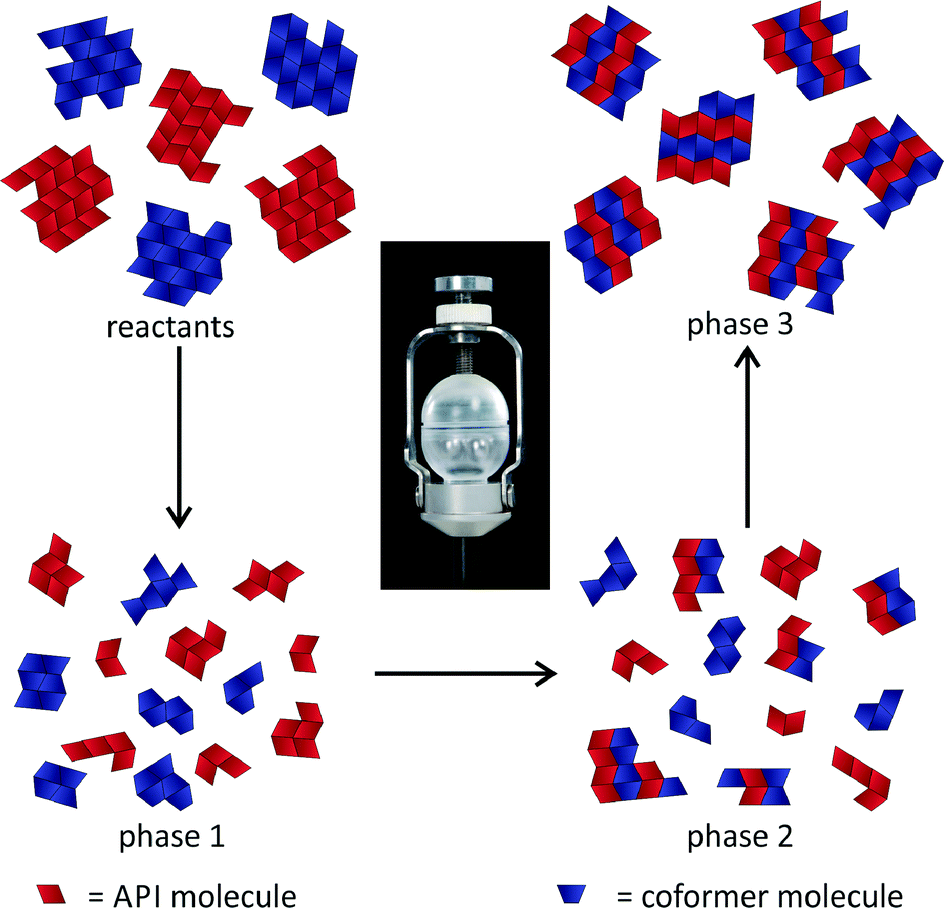 | ||
| Fig. 5 Representation of the mechanism during the milling synthesis of the 2:1 cocrystal of tb and ox. | ||
Experimental
Materials
Theobromine, C7H8N4O2, (99%, Acros Organics, Belgium) and oxalic acid dihydrate, C2H2O4·2H2O, (≥99+%, Acros Organics, Belgium) were purchased commercially and were used without further purification.Milling synthesis
The synthesis of the title compound was conducted by neat grinding in a ball mill (MM400, Retsch, Germany) at 30 Hz for 25 min in a molar ratio of 1.9:1 theobromine:oxalic acid dihydrate. A 10 mL steel vessel with two steel balls (10 mm) was used for a total load of 1 g.
XRD measurements
The obtained product was investigated by PXRD. The obtained powder pattern did not show any residues of the reagents. All X-ray diffraction experiments were carried out using a D8 diffractometer (Bruker AXS, Karlsruhe, Germany) in transmission geometry (Cu-Kα1 radiation, λ = 1.54056 Å). The structure was solved based on the PXRD pattern using the open source program FOX for indexing and calculating the structure.42 The program CHEKCELL was used to confirm the unit cell and the space group.43 FOX uses global optimisation algorithms to solve the structure by performing trials in direct space. This search algorithm uses random sampling coupled with simulated temperature annealing to locate the global minimum of the figure-of-merit factor. To reduce the total number of degrees of freedom, the theobromine molecule was treated as rigid. The crystal structure of the cocrystal was solved by the simulated annealing procedure on a standard personal computer within 12 h finding the deepest minimum of the cost function several times during the procedure. To complete the structure determination, the structural solution obtained from Monte Carlo/simulated annealing was subsequently subjected to Rietveld refinement employing the TOPAS software.44 CCDC 1028891 contains the supplementary crystallographic data for the tb:ox 2:1 cocrystal.
Synchrotron measurements
In situ measurements were performed at the microfocus beamline μSpot (BESSY II, Helmholtz Centre Berlin for Materials and Energy, Germany) in transmission geometry. The powder patterns were collected at a wavelength of 1.0000 Å using a Si (111) double-crystal monochromator. A two-dimensional MarMosaic CCD X-ray detector with 3072 × 3072 pixels was used to record the scattering intensity. In a typical experiment, the XRD patterns were collected every 10 s. The obtained scattering images were processed and converted into diagrams of scattered intensities versus the scattering vector q (q = 4π/λ sinθ) employing an algorithm from the FIT2D software.45 For the graphical representations, the q values were transformed into the diffraction angle 2θ (Cu) to provide a direct comparison to the results obtained by XRD experiments performed with Cu radiation. The in situ monitoring of the synthesis of the title compound was conducted by neat grinding in a Mini-Mill PULVERISETTE 23 (Fritsch, Germany) at 30 Hz for 20 min in a molar ratio of 1.9:1 theobromine:oxalic acid dihydrate. A 10 mL self-constructed Perspex vessel with two steel balls (10 mm) was used for a total load of 1 g. Every 30 s of milling a powder pattern of the sample was taken.
Raman spectroscopy
Raman measurements were performed on a Raman RXN1™ analyser (Kaiser Optical Systems, France). The spectra were collected using a laser with a wavelength of λ = 785 nm and a contactless probe head (working distance: 1.5 cm, spot size: 1.0 mm). The Raman spectra were recorded with an acquisition time of 5 s and 5 accumulations. NIR excitation radiation at λ = 785 nm and irradiation of 6.6 W cm−2 were performed.ssNMR spectroscopy
1H magic angle spinning (MAS) NMR spectra were recorded on a Bruker AVANCE 400 spectrometer using a 2.5 mm double-bearing MAS probe (Bruker BioSpin), applying a spinning speed of 20 kHz. The 1H MAS NMR spectra were recorded with a π/2 pulse length of 3.6 μs, a recycle delay of 5 s and an accumulation number of 256. Existent background signals were suppressed with a phase-cycled depth pulse sequence according to Cory and Ritchey.46DTA and TGA measurements
DTA and TGA measurements were conducted using a thermobalance SETARAM TAG24 in 1600 °C equipment. The measurements were performed in an open Pt jar under N2/synthetic air flow with a heating rate of 10 K min−1. No cycle measurements were taken.Conclusions
The crystal structure of a tb cocrystal with ox in a 2:1 ratio was solved from powder diffraction data. The cocrystal was synthesised mechanochemically. Due to the extremely poor solubility of tb this cocrystal could not be obtained from solution. Based on the Raman spectroscopy and ssNMR data the formation of a salt could be excluded. The synthesis pathway was investigated using in situ XRD and a three step mechanism was derived. The experiment proved that this approach is feasible for the characterisation of mechanochemical reactions.
Acknowledgements
We are grateful to Dr. S. Reinsch (BAM) for DTA-TGA measurements.Notes and references
- O. Almarsson and M. J. Zaworotko, Chem. Commun., 2004, 1889–1896 RSC.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. Rodríguez-Hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. Kumar Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147–2152 CAS.
- H. D. Clarke, K. K. Arora, H. Bass, P. Kavuru, T. T. Ong, T. Pujari, L. Wojtas and M. J. Zaworotko, Cryst. Growth Des., 2010, 10, 2152–2167 CAS.
- N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950–2967 CAS.
- B. Sarma, J. Chen, H. Y. Hsi and A. S. Myerson, Korean J. Chem. Eng., 2011, 28, 315–322 CrossRef CAS.
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris, Pharm. Res., 2001, 18, 859–866 CrossRef CAS.
- J.-P. Brog, C.-L. Chanez, A. Crochet and K. M. Fromm, RSC Adv., 2013, 3, 16905–16931 RSC.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed.
- Y. Umeda, T. Fukami, T. Furuishi, T. Suzuki, K. Tanjoh and K. Tomono, Drug Dev. Ind. Pharm., 2009, 35, 843–851 CrossRef CAS PubMed.
- A. T. M. Serajuddin, Adv. Drug Delivery Rev., 2007, 59, 603–616 CrossRef CAS PubMed.
- R. Chadha, A. Saini, P. Arora and S. Bhandari, Crit. Rev. Ther. Drug Carrier Syst., 2012, 29, 183–218 CrossRef CAS.
- C. B. Aakeroy, M. E. Fasulo and J. Desper, Mol. Pharmaceutics, 2007, 4, 317–322 CrossRef PubMed.
- M. C. Etter and G. M. Frankenbach, Chem. Mater., 1989, 1, 10–12 CrossRef CAS.
- T. Friščić and W. Jones, J. Pharm. Pharmacol., 2010, 62, 1547–1559 CrossRef PubMed.
- C. B. Aakeröy, M. E. Fasulo and J. Desper, Mol. Pharmaceutics, 2007, 4, 317–322 CrossRef PubMed.
- M. J. Zaworotko, Cryst. Growth Des., 2006, 7, 4–9 Search PubMed.
- D. P. McNamara, S. L. Childs, J. Giordano, A. Iarriccio, J. Cassidy, M. S. Shet, R. Mannion, E. O'Donnell and A. Park, Pharm. Res., 2006, 23, 1888–1897 CrossRef CAS PubMed.
- D. J. Good and N. R. Rodríguez-Hornedo, Cryst. Growth Des., 2009, 9, 2252–2264 CAS.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Int. J. Pharm., 2006, 320, 114–123 CrossRef CAS PubMed.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1013–1021 CAS.
- N. Chieng, M. Hubert, D. Saville, T. Rades and J. Aaltonen, Cryst. Growth Des., 2009, 9, 2377–2386 CAS.
- J. S. Stevens, S. J. Byard, C. A. Muryn and S. L. M. Schroeder, J. Phys. Chem. B, 2010, 114, 13961–13969 CrossRef CAS PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. Harris, G. Hyett and W. Jones, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- D. Braga, L. Maini, M. Polito and F. Grepioni, Chem. Commun., 2002, 2302–2303 RSC.
- S. Karki, T. Friscic and W. Jones, CrystEngComm, 2009, 11, 470–481 RSC.
- V. R. Pedireddi, W. Jones, A. P. Chorlton and R. Docherty, Chem. Commun., 1996, 987–988 RSC.
- M. R. Caira, L. R. Nassimbeni and A. F. Wildervanck, J. Chem. Soc., Perkin Trans. 2, 1995, 2213–2216 RSC.
- M. C. Etter, S. M. Reutzel and C. G. Choo, J. Am. Chem. Soc., 1993, 115, 4411–4412 CrossRef CAS.
- T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimäki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66–73 CrossRef PubMed.
- I. Halasz, S. A. J. Kimber, P. J. Beldon, A. M. Belenguer, F. Adams, V. Honkimäki, R. C. Nightingale, R. E. Dinnebier and T. Friščić, Nat. Protoc., 2013, 8, 1718–1729 CrossRef PubMed.
- I. Halasz, A. Puškarić, S. A. J. Kimber, P. J. Beldon, A. M. Belenguer, F. Adams, V. Honkimäki, R. E. Dinnebier, B. Patel, W. Jones, V. Štrukil and T. Friščić, Angew. Chem., Int. Ed., 2013, 52, 11538–11541 CrossRef CAS PubMed.
- S. Karki, L. Fábián, T. Friščić and W. Jones, Org. Lett., 2007, 9, 3133–3136 CrossRef CAS PubMed.
- N. Madusanka, M. D. Eddleston, M. Arhangelskis and W. Jones, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 72–80 CAS.
- J. I. Langford and D. Louer, Rep. Prog. Phys., 1996, 59, 131–234 CrossRef CAS.
- Y. Ebisuzaki and S. M. Angel, J. Raman Spectrosc., 1981, 11, 306–311 CrossRef CAS.
- L. Batzdorf, F. Fischer, M. Wilke, K. J. Wenzel and F. Emmerling, Angew. Chem., Int. Ed., 2014 DOI:10.1002/anie.201409834R1.
- P. Baláž, in Mechanochemistry in Nanoscience and Minerals Engineering, Springer, 2008, pp. 1–102 Search PubMed.
- F. P. Bowden and D. Tabor, The friction and lubrication of solids, Clarendon Press, Oxford, 1958 Search PubMed.
- F. P. Bowden and A. D. Yoffe, The Initiation and Growth of Explosion in Liquids and Solids, CUP Archive, 1952 Search PubMed.
- P. Thiessen, Abh. Dtsch. Akad. Wiss. Berlin, Kl. Chem., Geol. Biol., 1966, 15 CAS.
- X. Ma, W. Yuan, S. E. J. Bell and S. L. James, Chem. Commun., 2014, 50, 1585–1587 RSC.
- V. Favre-Nicolin and R. Cerny, J. Appl. Crystallogr., 2002, 35, 734–743 CrossRef CAS.
- J. Laugier and B. Bochu, Chekcell, 2001 Search PubMed.
- Topas Version 2.0, General Profile and Structure Analysis Software for Powder Diffraction Data (User Manual), Bruker AXS, Karlsruhe (Germany), 2000 Search PubMed.
- A. P. Hammersley, K. Brown, W. Burmeister, L. Claustre, A. Gonzalez, S. McSweeney, E. Mitchell, J.-P. Moy, S. O. Svensson and A. W. Thompson, J. Synchrotron Radiat., 1997, 4, 67–77 CrossRef CAS PubMed.
- D. G. Cory and W. M. Ritchey, J. Magn. Reson., 1988, 80, 128–132 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: DTA-TGA measurements of the reactants and mass spectrometry coupled with DTA-TGA measurements of the cocrystal. CCDC 1028891. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce02066a |
| This journal is © The Royal Society of Chemistry 2015 |