
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetrazoles: calcium oxalate crystal growth modifiers†
Calum J.
McMulkin
,
Massimiliano
Massi
and
Franca
Jones
*
Curtin University, Department of Chemistry, GPO Box U1987, Perth, WA, Australia 6845. E-mail: F.Jones@curtin.edu.au; Fax: +61 618 9266 4699; Tel: +61 618 9266 7677
First published on 20th February 2015
Abstract
Molecules containing tetrazole substituents have become of interest due to their being bioisosteres of carboxylic acids and like their carboxylate counterparts, tetrazolate anions have been able to affect the crystal growth of barium sulphate and calcium carbonate. In this proof of principle study, we show that this behaviour also extends to calcium oxalate and therefore opens the possibility of using tetrazole-based additives for investigating mineralization processes of human pathological relevance.
Introduction
Biomineralization encompasses many biological crystallization events including urolithiasis (the formation of stones in the urinary tract).1 Thus, the formation of kidney stones is an undesired biomineralization event within human bodies. Calcium oxalate, calcium phosphate and uric acid constitute 89% of renal calculus, commonly known as kidney stones. Calcium oxalate dominates the constituents of kidney stones at 70%.2 Calcium oxalate has three hydrate forms, with calcium oxalate monohydrate (COM, CaC2O4·H2O) being the thermodynamically stable phase. Calcium oxalate dihydrate (COD, CaC2O4·2H2O) and calcium oxalate trihydrate (COT, CaC2O4·3H2O) are the meta-stable phases. All three crystal phases can be found within kidney stones.The formation of kidney stones causes extreme pain in patients and currently the lifetime risk of developing a kidney stone is 10–15% in the developed world and up to 20–25% in the Middle East.3 The current best-practises in dealing with kidney stones are medical expulsive therapy (MET) and shock-wave lithotripsy (SWL) both of which are not methods of prevention but rather are aimed at spontaneous passage of formed ureteral calculi, and ultrasound degradation of the particles too big for passage. The best pharmaceutical treatments include the use of hydrochlorothiazide, chlorthalidone or indapamide which act as thiazide or thiazide-like diuretics and inhibit the kidneys ability to retain water; these drugs however have potential drawbacks associated with the risk of hypotension (low blood-pressure) and hypokalemia (potassium depletion).3,4 Not surprisingly, major effort has been devoted to controlling the size and shape of the calcium oxalate with the aim of finding better strategies to deal with this condition.
Crystal growth modifiers are additives that can be used to gain control over crystallization processes, either in the form of promotion or inhibition. To date, a variety of additive types including; calixarenes,5,6 glycosaminoglycans,7 double-hydrophilic block copolymers,8 phospholipid monolayers,9 amino acids, proteins and carboxylic acids have been shown to impact the crystallization of calcium oxalate.10 Our previous work investigated the use of tetrazoles as crystal growth modifiers for inorganic solids. We showed that both the crystallization of barium sulphate and calcium carbonate are impacted on by tetrazoles in a unique way compared to their parent carboxylic acids.11 In this manuscript, we report a proof of principle study to determine whether tetrazoles can impact on the crystallization of calcium oxalate and if they do so at comparable levels to carboxylic acids. Thus, this is a first step in determining whether tetrazoles might hold any potential for crystal growth modification of biologically important minerals.
The methods of synthesising tetrazoles has been extensively researched12–19 due to the multitude of applications of tetrazole-containing compounds, including the use of tetrazoles in materials science and medicinal chemistry.20,21 Tetrazoles have been increasingly used in medicine due to their bioisostere nature with carboxylic acids.22 Carboxylic acids and tetrazoles are isosteres due to their similar pKa values resulting from their dissociation (details on the pKa's calculated or known for the molecules investigated in this work can be found in Table S1 in the ESI†). Thus it has become possible to replace the carboxylic acid functional group with that of the tetrazole ring for medicinal applications such as anti-asthmatic and anti-cancer drugs.23,24 An important aspect of this replacement is due to the tetrazole being a more metabolically stable substituent thus making it less susceptible to biodegradation.25
Experimental
Tetrazole synthesis
Fig. 1 shows the structures of the tetrazole molecules and carboxylic acid equivalents investigated in this work. All of the tetrazoles were prepared following a previously reported procedure,19 involving the 1,3-dipolar cycloaddition of the corresponding nitrile with sodium azide (NaN3) in the presence of triethylammonium hydrochloride. The tetrazole-containing carboxylic acid (TzBaH2) was synthesised by selective hydrolysis in aqueous sodium hydroxide at room temperature from the corresponding methyl ester. | ||
| Fig. 1 The tetrazole-substituted additives and carboxylic acids investigated in this work: 5-phenyl-2H-tetrazole (TzPhH), 4-(2H-tetrazol-5-yl)benzaldehyde (TzPhaH), 4-(2H-tetrazol-5-yl)benzonitrile (TzPhCNH), 1,4-bis-(2H-tetrazol-5-yl)benzene (TzTzH2), benzoic acid (BaPhH), 4-formylbenzoic acid (BaPhaH), 4-cyanobenzoic acid (BaPhCNH), terephthalic acid (BaBaH2), 4-(2H-tetrazol-5-yl)benzoic acid (BaTzH2). | ||
Crystallization of calcium oxalate methodology
Stock solutions of the starting two reagents (calcium chloride and sodium oxalate) were prepared. To a flask (250 mL), calcium chloride (0.444 g, 0.004 mol) was added into ultrapure water (200 mL) to make the stock calcium chloride solution (0.02 mol L−1). Next, sodium oxalate (0.536 g, 0.004 mol) was added to ultrapure water (200 mL) in a 250 mL flask to make the stock sodium oxalate solution (0.02 mol L−1), with the use of a sonicator when necessary. Stock solutions of the modifying species (tetrazoles or carboxylic acids) were made up to 10 g L−1 concentrations, at pH 7 (adjusted to ~pH 7 by 1.0 M sodium hydroxide solution), ready for addition. To obtain the calcium oxalate crystals 300–500 μL of the calcium chloride solution was added to 20 mL glass vials as required. Cleaned glass discs with a radius of 0.5 cm were placed at the bottom of each vial to provide a removable crystal growth site. Next the required amount of modifier was added to the flask followed by the addition of ultrapure water, almost up to a final volume (20 mL). The solution was equilibrated in a water bath to the reaction temperature, 30 °C, before the addition of sodium oxalate solution (equimolar, 300–500 μL) to begin the reaction. The vials were left in the 30 °C water bath for 3 days before the glass disc from each sample vial was removed and the excess fluid adsorbed using tissue paper. The glass vial was then placed in a new, dry sample vial for analysis. All samples had a constant final volume of 20 mL and pH ~7. The final concentrations of calcium oxalate in the vials ranged between 1.0 × 10−4 M and 5.0 × 10−4 M, but the results for a final concentration of 0.30 mM are presented here. All the experiments and the number of repeats are given in the ESI† (Table S2).Scanning electron (SEM)
SEM was performed at the Curtin University Electron Microscope Facility using a Zeiss Evo 40XVP SEM using either secondary electron or back-scatter detectors. Energy-dispersive X-ray spectroscopy (EDX) was also employed in determining the qualitative composition of our samples. Each sample analysed by SEM was coated with either gold or platinum.Complexation experiments
Commercially obtained COM (AR grade) was added in excess (0.25 g, 1.71 mmol) to 50 mL vials with varying concentrations of organic additives (0–5 g L−1), filled with ultrapure water and left for 1 week at room temperature.After 1 week, the filtrate was collected by vacuum through 0.2 μm filter membranes for analysis. Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) analysis was recorded on a Varian Vista Axial CCD ICP-AES at Murdoch University's Centre for Marine and Freshwater Research Laboratory. The final measured calcium ion concentrations for these experiments can be found in Table S3 in the ESI.†
Measurement of size from SEM images
Each SEM image used in either the manuscript or supplementary information was run through the free software, ImageJ, to determine the number of particles, their size and standard deviation of particles from the mean. The measuring tool and scale bar calibration was used to measure the length (c-axis length) and width of each particle in the SEM image. Table S4 in the ESI† was generated from the collection of this data.Confocal raman spectroscopy
While XRD would have been the characterization method of choice to determine the different hydrates of calcium oxalate formed, the small amount of solids present meant this technique was not appropriate. Thus, Raman, which has also been shown to be specific to mineral form and chemistry, was used instead.26–29The Confocal Raman Spectroscopy (CRM) and Scanning Near-Field optical Microscopy (SNOM) measurements were obtained using the WITec alpha 300SAR utilising a frequency-doubled NdYAG laser of wavelength 532 nm and of 50 mW power. Each Raman spectrum collected consists of 10 accumulations at an integration time of 0.08371 seconds and a grating of 600 g mm−1. Analysis was performed using the WITec Project FOUR software.
Timed experiments
All conditions were the same as mentioned in the crystallization of calcium oxalate except that the samples were removed at different time periods up to a time of 1 month. The additives investigated were TzPh− (6.84 mM) and BaPh− (8.18 mM) as further investigation into the dissolution mechanism occurring in the presence of the phenyl-tetrazole species TzPh− was required.Turbidimetric methodology
Ultrapure water (3.48 mL) was added to a ~5 mL cuvette and a background reading was recorded before the addition of organic additive (0.4 mL), calcium chloride and sodium oxalate solutions (60 μL each) before the turbidity was monitored for thirty minutes. Turbidity was measured by using a UV-vis instrument (GBC Double beam UV/VIS Spectrophotometer 916) at 900 nm at an integration time of 0.5 seconds and slit width of 2.0 nm for all samples. All samples were filtered through a 0.1 μm membrane filter to remove any particles in the solution that would provide a seed for nucleation. The final concentration of calcium oxalate in the cuvettes was 3.0 × 10−4 M. Turbidity experiments were conducted multiple times and averaged to give the final curve for induction time determination.Results and discussion
Control calcium oxalate monohydrate
In order to determine the impacts of both the tetrazole containing and the carboxylate containing molecules many control samples were imaged and analysed. Fig. 2 shows an SEM of the typical control crystals with COM morphology. As previously mentioned, COM is the thermodynamically stable phase for calcium oxalate and thus would be the expected product after 72 hours. These particles were confirmed as COM by confocal Raman spectroscopy (see ESI,† Fig. S1 and S2). The mean length of the control particles was found to be approximately 8 μm in length. No fluorescence was observed in the presence of COM for all laser strengths applied. ESI† Fig. S3 illustrates the crystal face indexing for COM used throughout this manuscript.30 | ||
| Fig. 2 SEM image of calcium oxalate monohydrate control particles (0.30 mM). | ||
Impact of carboxylic acid derivatives as compared to tetrazole derivatives on calcium oxalate crystallization
Fig. 3 presents the comparison between some of the carboxylic acid and tetrazole additives on calcium oxalate crystallization in this study. The impact of tetrazole additives are all shown on the left side of Fig. 3, while their carboxylic acid equivalent groups are shown on the right hand side. Starting from the top, both additives with an aldehyde functional group; Fig. 3a (TzPha−) and b (BaPha−) formed calcium oxalate crystals with shortened crystal lengths in the <001> direction (c-axis, see Fig. S3† for a schematic of the crystal directions) compared to the control samples previously seen. Upon close inspection, some crystals (Fig. 3a) have the {010} face missing and/or twinning is reduced resulting in some cube shaped crystals (see Fig. S4† for a magnified view of Fig. 3a). While both additives have modified the crystallization of calcium oxalate, the TzPha− additive was found to have altered the crystal shape and increased the average particle length to ~16.5 μm (see Table S4 in ESI†). BaPha−, on the other hand, decreased the average particle length compared to the control. | ||
| Fig. 3 SEM images of calcium oxalate (0.30 mM) formed in the presence of (a) 1.0 g L−1 (5.74 mM) TzPha−, (b) 1.0 g L−1 (6.66 mM) BaPha−, (c) 1.0 g L−1 (4.66 mM) TzTz2−, (d) 1.0 g L−1 (6.02 mM) BaBa2−, (e) 1.0 g L−1 (6.84 mM) TzPh− and (f) 1.0 g L−1 (8.18 mM) BaPh−. | ||
The addition of TzTz2− resulted in many crystals with shortened a-axis lengths (Fig. 3c), meaning the crystal thickness was reduced, whilst the presence of BaBa2− (Fig. 3d) resulted in crystals more than twice the size of the control samples (see Table S4†). The final images show calcium oxalate crystals in the presence of TzPh− and BaPh− (Fig. 3e and f, respectively). The tetrazole species has impacted the COM crystals by showing some re-dissolution of the crystal centre on the {100} face through the entire crystal width. Investigating the possible means by which the tetrazole does this is discussed later. The carboxylic acid additive on the other hand, showed no signs of this re-dissolution and did not show significant signs of morphological impact or alteration of the aggregation state of the crystals. Both species TzPh− and BaPh− had similar gross morphology and mean particle sizes (Table S4†) indicating that preferential adsorption onto specific crystal faces does not appear to be occurring.
The species, TzBa2− showed only weak signs of inhibiting calcium oxalate crystallization (see Fig. 4 and S5†) and these were determined to be COM by Raman, unlike some carboxylic acids that also change the hydrate formed.31,32 The morphology of those particles that were not broken do not appear to be significantly different to the control particles, and the average length of the particles in the presence of this additive was found to be ~6 μm compared to the control length of ~7 μm, which is within the standard deviation of the measurements.
 | ||
| Fig. 4 SEM image of calcium oxalate (0.30 mM) particles formed in the presence of 5 g L−1 TzBa2− (0.0301 M). | ||
The presence of TzPhCN− (Fig. 5a) showed a similar action to that when BaPhCN− (Fig. 5b) additive was present, the additives appearing to have a significant impact on growth in the <100> (the thickness of the crystals). In the presence of both these additives the calcium oxalate crystals formed are significantly thinner than the control crystals. The presence of TzPhCN− appears to have a greater impact than BaPhCN− on the size as confirmed by the quantitative data (see Table S4†). At the lower concentration of 1 g L−1 of additive, the COM particles are almost indistinguishable from the control particles (see Fig. S6 and S7†).
 | ||
| Fig. 5 SEM image of calcium oxalate (0.30 mM) particles formed in the presence of 5 g L−1 a) TzPhCN− (0.029 M) and b) BaPhCN− (0.034 M). | ||
Turbidity
To better determine the impact of these small molecules on the nucleation behaviour of calcium oxalate, we undertook turbidimetric studies to assess their impact on homogenous nucleation. Turbidity was used to monitor the onset of nucleation and the induction time from these experiments determined. The important parameter in these studies is not the turbidity (which is impacted by many factors such as size, shape and number of particles)33,34 but the induction time, which is the time the turbidity increases above baseline.35 Induction time is inversely related to the homogenous nucleation rate. Increasing induction time suggests that the organic molecules are adsorbing onto the nuclei and changing their surface free energy (provided supersaturation is not significantly altered).36 Due to the small volumes and low concentrations used in this study, there was scatter in the turbidimetric data, thus 4–6 data sets were averaged to minimise these errors. The complete set of turbidity curves can be found in the supplementary information along with the induction time shown by an arrow (Fig. S8–S16†). The most important information, however, was how the presence of the additives altered the nucleation rate compared to the control, thus, trends being more important than actual values. The control sample (Fig. S14†) showed an induction time of between 25–35 seconds based on the time required for the turbidity to rise above the background. When the additive TzTz2− (Fig. S15†) was present during crystallization the induction time was found to be ~10 seconds. In comparison the presence of BaBa2− (Fig. S16†) was found to have an even shorter induction time, almost occurring instantaneously, at ~5 seconds.None of the tetrazole additives showed a significant lengthening of the induction time. In fact for most of the tetrazole additives the opposite was found. Two tetrazoles, TzPh− and TzPha−, showed similar induction times to the control. In addition, this result suggests that despite the fact that the doubly charged additives complex more with calcium in solution (see section below on solution behaviour) they do not lower the nucleation rate of calcium oxalate monohydrate. For the carboxylate molecules, BaPh−, BaPhCN− and BaPha−, each was found to promote nucleation somewhat with induction times of 15–25 seconds, 5–15 and 10–15 seconds, respectively. Similar to the tetrazoles then, the carboxylic acid derivatives appear to promote nucleation slightly.
The ability of the tetrazoles to complex calcium was investigated to determine whether the modifiers altered supersaturation significantly and whether complexation could lead to dissolution of the formed COM particles.
Complexation behaviour
The ability of the various molecules to complex calcium was undertaken by measuring the dissolution of commercially obtained COM with the six inhibitors highlighted in Fig. 6, in ultrapure water. The calcium ion content after a week was analysed by ICP-AES, which determined that increased solubility by complexation did not significantly occur for many of the molecules (Fig. 6), in particular in the presence of TzPh−. However, it was shown from this data set that the TzTz2−, BaBa2− and TzBa2− additives showed significant increases in the dissolution of calcium oxalate monohydrate compared to the other additives analysed (see Table S3† for concentration information). The molecules containing more than one functional group, therefore, would change the supersaturation solutions and may account for the observed morphological impacts in the presence of these additives.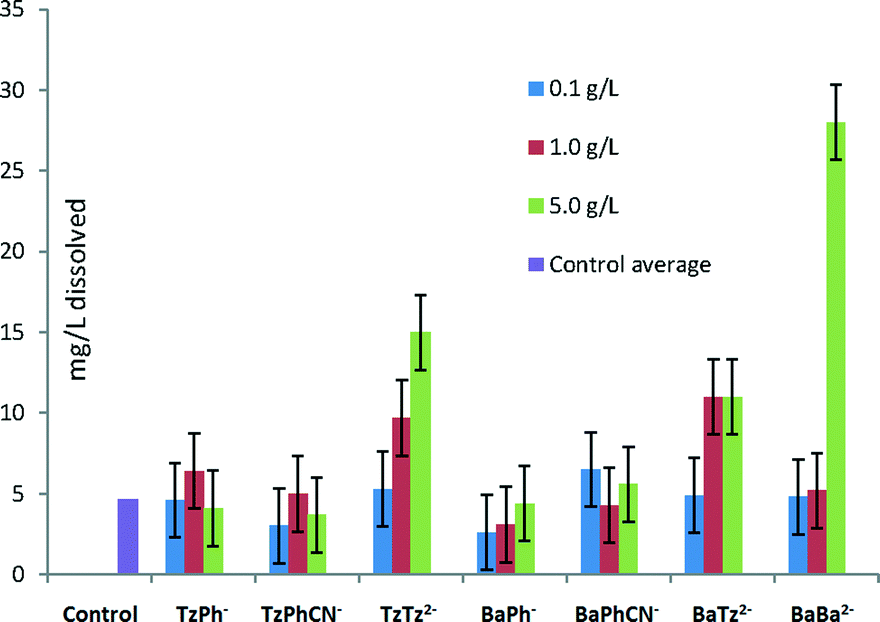 | ||
| Fig. 6 Dissolution of Ca2+ (mg L−1) from COM in the presence of additive species in solution measured by ICP-AES. | ||
In addition, since the tetrazole, TzPh−, does not significantly complex calcium the re-dissolution does not appear to be due to subsequent complexation and dissolution.
Timed experiments
Prolonged timed experiments were conducted to determine if,i) in the presence of the additive TzPh−, the crystals grew in the manner they are observed and
ii) after the COM dissolved whether some other solid phase re-precipitated.
Initially, the majority of particles formed were the thermodynamically stable COM (see Fig. S17a†) having a morphology that is also equivalent to the control particles. Particles larger in mean length than an average control sample were obtained according to the quantitative data; standard COM control crystals usually averaged ~7 μm in length while in the presence of the tetrazole average lengths from 9–22 μm were observed in different repeats. After >3 days dissolution of the interior of the particles can be observed (as shown in Fig. S17b†) and is a reproducible affect seen in Fig. 3e, S18 and S19.†
No evidence of calcium oxalate re-precipitation is observed of any form. After 19 days few particles with COM shape remained, and there is microbial action evident as the laboratory conditions were not sterile. This issue persisted even when we attempted to sterilise by filtration (filtering all solutions using a 0.2 μm pore membrane). Experiments with azide are currently underway. The timed experiments did show that the crystals did not grow in a hollowed manner from the beginning but were in fact similar to control crystals, however we were not able to determine any recrystallization due to the bacterial growth. This bacterial growth did not occur for the control experiment, nor in the presence of the carboxylate containing molecule even after 19 days (see Fig. S20a, b and c†).
Using Confocal Raman spectroscopy, the crystals taken after 3 days in the presence of the tetrazole; still in typical COM shape with no visible signs of hollowing were analysed. These particles showed a Raman spectrum (Fig. S21a†) with a significant degree of fluorescence in the background. Further investigation found that this fluorescence only occurred in the presence of the tetrazole molecule. In addition, the two main identified peaks at ~1350 cm−1 and ~1622 cm−1 are believed to be the tetrazole peaks (Fig. S21b†) but modified on adsorption (for example, due to de-protonation).37 The unadsorbed tetrazole has these peaks at 1411 cm−1 and 1602 cm−1. In any event, these bands are different to the control COM crystal's Raman spectrum (Fig. S21c†), showing the expected doublet for the monohydrate at 1460, 1480 cm−1 and which does not emit such broad fluorescence as seen in Fig. S21a.†
This implies that the tetrazole species is present in the crystal prior to re-dissolution. The data thus far suggests it is the incorporation of the tetrazole that causes this re-dissolution by weakening the crystal structure at defects or areas of strain.
Conclusions
From this study we have shown that tetrazoles can impact on COM morphology and size significantly. Of the molecules investigated the TzPhCN−, TzTz2− and TzPha−, were found to be the most potent. The growth directions most impacted by the tetrazoles are the <100> and <001> of COM. In all cases we found COM was formed in the presence of these tetrazoles, not COD, and this is thought to be due to the pH of the experiments falling between 6.5 and 7.5, while COD has been shown to form favourably in basic conditions.38 The tetrazole molecules were generally found to promote homogenous nucleation.The tetrazole additives throughout this study have been shown to impact more than their carboxylic acid equivalent groups (as lower concentrations are required to achieve similar or more significant morphological impacts) across a variety of functional groups. The increase in the number of functional groups from one to two tetrazole or carboxylic acid groups is seen to increase their impact on calcium oxalate crystallization as has been observed for other systems.11
The re-dissolution observed in the presence of TzPh− was investigated in more detail. It was found that this tetrazole does not complex the calcium ion to any significant extent but is present with the COM prior to the dissolution occurring. The timed experiments showed that the COM doesn't grow in this fashion and that therefore the most likely mechanism is that the tetrazole is incorporated in the crystal and that the dissolution is due to this incorporation weakening the crystal structure. This is further being investigated due to microbial action interfering with the longer-term experiments.
In conclusion, it can be stated that tetrazoles can be used as crystal growth modifiers of calcium oxalate monohydrate. Furthermore, we plan to continue research in this area to explore a variety of organic backbones, different functional groups and their stereochemistry. It will be interesting to investigate the differences in stereoisomers in the ortho and meta substituted positions in addition to further investigations into functional groups that are electronegative and electropositive; this is due to the most significant modifiers being identified, in this study, as those that are electron withdrawing. Experiments closer to physiological conditions and biologically relevant environments are also envisaged to ascertain whether the ability of the tetrazole to impact crystallization is retained.
Acknowledgements
The authors acknowledge the use of equipment and technical assistance of the Curtin University Electron Microscope Facility, which has been partially funded by the University, state and Commonwealth Governments.References
- M. S. Pearle, E. A. Calhoun and G. C. Curhan, in Urological Diseases in America, 2007, pp. 283–319 Search PubMed.
- L. Smith, Trans. Am. Clin. Climatol. Assoc., 2002, 113, 1–20 Search PubMed.
- O. W. Moe, Lancet, 2006, 367, 333–344 CrossRef CAS.
- O. W. Moe, M. S. Pearle and K. Sakhaee, Kidney Int., 2011, 79, 385–392 CrossRef CAS PubMed.
- F. Jones, M. Mocerino, M. I. Ogden, A. Oliveira and G. M. Parkinson, Cryst. Growth Des., 2005, 5, 2336–2343 CAS.
- M. J. Bartlett, M. Mocerino, M. I. Ogden, A. Oliveira, G. M. Parkinson, J. K. Petterson and M. M. Reyhani, J. Mater. Sci. Technol., 2005, 21, 1–5 Search PubMed.
- Y. Shirane, Y. Kurokawa, S. Miyashita, H. Komatsu and S. Kagawa, Urol. Res., 1999, 27, 426–431 CrossRef CAS.
- D. Zhang, L. Qi, J. Ma and H. Cheng, Chem. Mater., 2002, 2450–2457 CrossRef CAS.
- S. R. Letellier, M. J. Lochhead, A. A. Campbell and V. Vogel, Biochim. Biophys. Acta, 1998, 1380, 31–45 CrossRef CAS.
- P. Sayan, S. T. Sargut and B. Kıran, Cryst. Res. Technol., 2009, 44, 807–817 CrossRef CAS.
- M. Massi, M. I. Ogden, T. Radomirovic and F. Jones, CrystEngComm, 2010, 12, 4205 RSC.
- F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2002, 124, 12210–12216 CrossRef CAS PubMed.
- F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2003, 125, 9983–9987 CrossRef CAS PubMed.
- Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945–7950 CrossRef CAS PubMed.
- A. N. Chermahini, A. Teimouri and A. Moaddeli, Heteroat. Chem., 2011, 22, 168–173 CrossRef CAS.
- A. Teimouri and A. Najafi Chermahini, Polyhedron, 2011, 30, 2606–2610 CrossRef CAS PubMed.
- R. Huisgen and H. Markgraf, Chem. Ber., 1960, 93, 2106–2124 CrossRef CAS.
- Y. Zhu, Y. Ren and C. Cai, Helv. Chim. Acta, 2009, 92, 171–175 CrossRef CAS.
- K. Koguro, T. Oga, S. Mitsui and R. Orita, Synthesis, 1998, 910–914 CrossRef CAS PubMed.
- S. Stagni, S. Colella, A. Palazzi, G. Valenti, S. Zacchini, F. Paolucci, M. Marcaccio, R. Q. Albuquerque and L. De Cola, Inorg. Chem., 2008, 47, 10509–10521 CrossRef CAS PubMed.
- D. D'Alessio, S. Muzzioli, B. W. Skelton, S. Stagni, M. Massi and M. I. Ogden, Dalton Trans., 2012, 41, 4736–4739 RSC.
- R. J. Herr, Bioorg. Med. Chem., 2002, 10, 3379–3393 CrossRef CAS.
- P. I. Eacho, P. S. Foxworthy, R. D. Dillard, C. A. Whitesitt, D. K. Herron and W. S. Marshall, Toxicol. Appl. Pharmacol., 1989, 100, 177–184 CrossRef CAS.
- J. H. Fleisch, M. L. Cloud and W. S. Marshall, Ann. N. Y. Acad. Sci., 1988, 524, 356–368 CrossRef CAS PubMed.
- S. J. Wittenberger, Org. Prep. Proced. Int., 2009, 26, 499–531 CrossRef.
- H. G. M. Edwards, D. W. Farwell and R. Jenkins, J. Raman Spectrosc., 1992, 23, 185–189 CrossRef CAS.
- A. T. Tu, J. Chin. Chem. Soc., 2003, 50, 1–10 CAS.
- J. R. Guerra-López, J. A. Güida and C. O. Della Védova, Urol. Res., 2010, 38, 383–390 CrossRef PubMed.
- C. Paluszkiewicz, M. Gałka, W. Kwiatek, A. Parczewski and S. Walas, Biospectroscopy, 1997, 3, 403–407 CrossRef CAS.
- V. Tazzoli and C. Domeneghetti, Am. Mineral., 1980, 65, 327–334 CAS.
- J. Ouyang, L. Duan, B. Tieke and D. Ko, Langmuir, 2003, 8980–8985 CrossRef CAS.
- S. Deng and J. Ouyang, Chin. J. Chem., 2007, 25, 1379–1384 CrossRef CAS.
- L. Lafferrere, C. Hoff and S. Veesler, Cryst. Growth Des., 2004, 4, 1175–1180 CAS.
- L. Smith, K. Roberts, D. Machin and G. McLeod, J. Cryst. Growth, 2001, 226, 158–167 CrossRef CAS.
- A. Kuldipkumar, G. S. Kwon and G. G. Z. Zhang, Cryst. Growth Des., 2007, 7, 234–242 CAS.
- M. C. van der Leeden, D. Kashchiev and G. M. van Rosmalen, J. Cryst. Growth, 1993, 130, 221–232 CrossRef CAS.
- E.-S. M. Sherif, R. M. Erasmus and J. D. Comins, Corros. Sci., 2008, 50, 3439–3445 CrossRef CAS PubMed.
- V. Thongboonkerd, T. Semangoen and S. Chutipongtanate, Clin. Chim. Acta, 2006, 367, 120–131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images, turbidity spectra, and Raman spectra of calcium oxalate particles in the presence of tetrazole and carboxylate additives. Tables of quantitative data extracted from SEM images, dissolution experiment concentration information and additive pKa values. See DOI: 10.1039/c4ce01717j |
| This journal is © The Royal Society of Chemistry 2015 |