
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure-directing factors when introducing hydrogen bond functionality to metal–organic frameworks†
Ross S.
Forgan
*a,
Ross J.
Marshall
a,
Mona
Struckmann
a,
Aurore B.
Bleine
a,
De-Liang
Long
a,
María C.
Bernini
b and
David
Fairen-Jimenez
 *c
*c
aSchool of Chemistry, University of Glasgow, Joseph Black Building, University Avenue, Glasgow, G12 8QQ, UK. E-mail: ross.forgan@glasgow.ac.uk; Web: http://www.forganlab.com
bFacultad de Química, Bioquímica y Farmacia, Universidad Nacional de San Luis, Instituto de Tecnología Química-CONICET, San Luis, Argentina
cDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Pembroke St., Cambridge CB2 3RA, UK. E-mail: df334@cam.ac.uk; Web: http://people.ds.cam.ac.uk/df334
First published on 24th October 2014
Abstract
The introduction of H-bond donor/acceptor functionality into metal–organic frameworks (MOFs) can have a beneficial effect on their molecular recognition, uptake selectivity and catalytic properties. The changes in ligand geometry induced by incorporation of functional groups may also affect the topology and composition of the resultant MOFs. Herein, we present a comprehensive study of functional group incorporation into MOFs, linked by either Zn2+ paddlewheel units or monomeric Zn2+ corners, which exhibit pcu and dia topology, respectively. Crystallographic analysis shows that amide groups can be easily incorporated into isoreticular pcu pillared-MOFs, whilst integration of urea units results in materials with dia topology. Molecular simulations allow the examination of hypothetical structures with differing constitutions and topologies, and highlight the influence of the urea units in generating the experimentally observed topologies. Noncovalent interactions between independent nets may be significant structure-directing influences, a finding which has great implications for the design of MOFs containing more complex functional groups.
1. Introduction
Metal–organic frameworks (MOFs),1 crystalline network structures comprised of multitopic organic ligands, or struts, linked by metal ions or metal clusters, have found application in many areas as a result of their tunable functionality and porosity, including, but not limited to, gas storage,2 molecular separations,3 catalysis,4 drug delivery5 and sensing.6 Enhancement of desired properties is often achieved through functionalization of the MOF, either by post-synthetic modification7 or by installing functional groups into the constituent ligands prior to MOF assembly. Pendant nucleophilic groups, such as –NH2 and OH moieties, are known to enhance8 CO2 capture through strong interactions with the quadrupolar moment of the gaseous substrate, and, in some cases, through formation9 of transient covalent bonds between the MOF and the guest. Amide moieties incorporated into MOF struts give a convenient synthetic route to larger linkers, and are known10 to enhance CO2 capture through favourable H-bonding and dipole–quadrupole interactions. A diverse range of amide-containing MOFs has been prepared11 in the last few years to take advantage of this improved affinity for CO2. Incorporation12 of urea units into MOFs generates heterogeneous organocatalytic species, which act as H-bond donor centres to the substrate(s) in question.Incorporation of hydrogen bond functionality directly into MOF struts is clearly an attractive method for tuning and enhancing desirable properties, and an isoreticular series of frameworks containing differing functional groups would offer insight into the individual effect of each moiety on properties such as gas uptake and molecular recognition. Amide and urea units, however, bring the added complication of altering ligand geometry when compared to unsubstituted control ligands, with deviations of the disposition of the donor units from linearity that may influence the structure of the resultant MOFs.
In order to examine the effect of introducing hydrogen bonding units into MOFs on their topologies in a sequential manner, we have initially chosen the family of so-called pillared13 MOFs – structures comprised of two dimensional sheets of diacids linked by dimeric Zn(II)-paddlewheel secondary building units (SBUs) that are connected in the third dimension by nitrogen donor pillars in the pcu topology – as a system that allows the sequential incorporation of increasing numbers of functional groups. A series of functionalised diacid ligands (L1·2H–L3·2H) and pillars (P1–P3) has been collated (Fig. 1a) and, using the known14 compound [Zn2(L1)2(P1)]n as a basic starting point (Fig. 1b), the structures of four new resultant MOFs are reported. Of these structures, two of them show a pcu topology, whereas the two other structures, obtained with a pillar (P3) that includes the urea unit, exhibit a dia topology. In addition to these four materials obtained experimentally, we have built a series of new hypothetical structures of varying compositions and topologies in order to analyse the geometries and interactions between ligands and pillars present in both pcu and dia topologies. Density functional theory calculations have been used previously to examine the energy landscapes of MOFs15 as well as their physical properties.16 However, since these calculations are computationally expensive, we followed17 a classical simulation approach that allowed us to easily obtain the new hypothetical constructs and examine their structures.
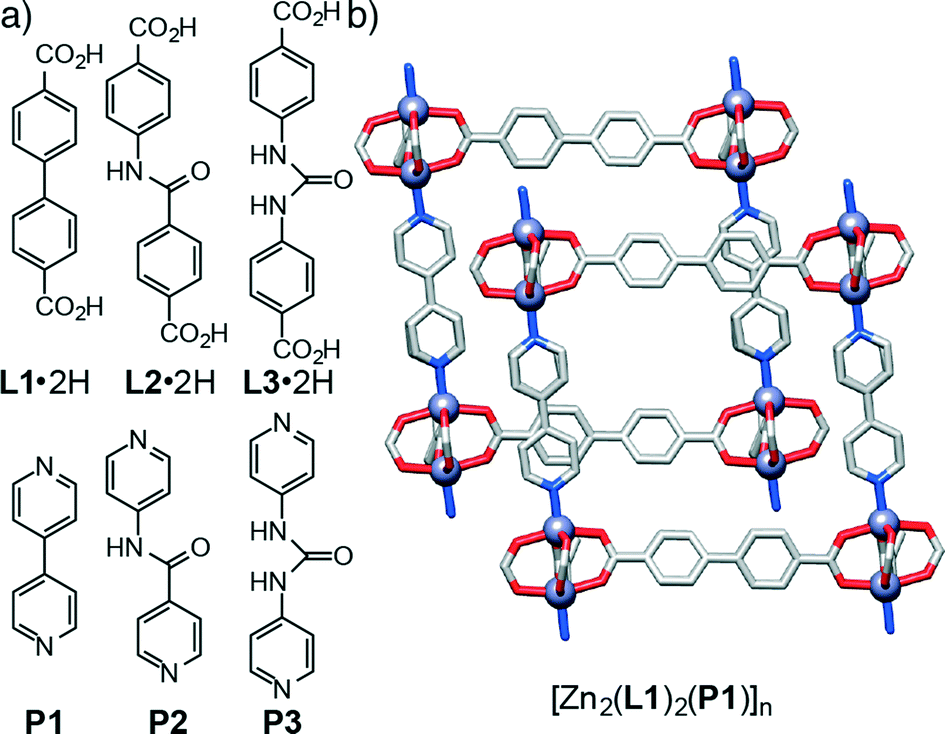 | ||
| Fig. 1 a) Structures of the diacid ligands (L1·2H–L3·2H) and di-pyridyl pillars (P1–P3) examined in this study. b) Portion of the solid-state structure of [Zn2(L1)2(P1)]n, the basic framework upon which the work is based. Redrawn from CCDC deposition FEFCUQ with hydrogen atoms and disorder omitted for clarity. | ||
2. Results and discussion
In addition to the known structure [Zn2(L1)2(P1)]n, four further MOFs were isolated18 after solvothermal treatment (see experimental section) of mixtures of [Zn(NO3)2]·6H2O, a diacid ligand and a pillar in N,N′-dimethylformamide (DMF), and their structures elucidated by single crystal X-ray diffraction (Table 1). While the first two structures using the unfunctionalised and monoamide functionalised ligands and pillars showed the same Zn-paddlewheel units and pcu topology as the parent [Zn2(L1)2(P1)]n precursor, the two other structures, using the urea-functionalised P3 pillar, showed a monomeric Zn2+ metal corner and a change into a dia topology.| Compound 1 | Compound 2 | Compound 3 | Compound 4 | |
|---|---|---|---|---|
| a Twin abs correction applied. | ||||
| Formula | C57H67N9O15Zn2 | C64H82N12O18Zn2 | C37H46N8O9Zn | C38H47N9O10Zn |
| Mass (FM) | 1248.93 | 1438.15 | 812.19 | 855.21 |
| Crystal size (mm) | 0.408 × 0.356 × 0.142 | 0.536 × 0.262 × 0.145 | 0.100 × 0.050 × 0.040 | 0.110 × 0.080 × 0.050 |
| T (K) | 150(2) | 150(2) | 150(2) | 150(2) |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | P21/n | P21/c |
| a (Å) | 15.1431(6) | 19.7220(5) | 16.381(3) | 14.529(2) |
| b (Å) | 15.2322(6) | 28.0588(6) | 14.170(3) | 16.543(2) |
| c (Å) | 16.0597(4) | 28.0835(4) | 19.415(4) | 21.125(3) |
| α (°) | 98.484(2) | 90 | 90 | 90 |
| β (°) | 102.682(2) | 94.423(2) | 104.25(3) | 104.305(9) |
| γ (°) | 104.206(3) | 90 | 90 | 90 |
| V (Å3) | 3423.1(2) | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 494.4(6) 494.4(6) |
4367.9(16) | 4919.8(12) |
| Z | 2 | 8 | 4 | 4 |
| ρ (g cm−3) | 1.212 | 1.233 | 1.235 | 1.155 |
| F(000) | 1304 | 6032 | 1704 | 1792 |
| θ range (°) | 2.818–25.772 | 2.880–26.000 | 1.926–25.764 | 1.971–25.132 |
| Reflections | 54284 |
125275 |
70868 |
26904 |
| Independent reflections | 13023 |
30417 |
8546 | 12472 |
| R int | 0.0360 | 0.0624 | 0.0960a | 0.0876a |
| GOOF (F2) | 1.062 | 1.054 | 1.086 | 1.098 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0662 | R 1 = 0.0812 | R 1 = 0.0905 | R 1 = 0.1026 |
| wR2 = 0.2062 | wR2 = 0.2480 | wR2 = 0.2450 | wR2 = 0.2844 | |
| Final R indices (all data) | R 1 = 0.0759 | R 1 = 0.1393 | R 1 = 0.1336 | R 1 = 0.1688 |
| wR2 = 0.2157 | wR2 = 0.2952 | wR2 = 0.2956 | wR2 = 0.3282 | |
The first compound, 1, [Zn2(L1)2(P2)]n, forms through the combination of the unsubstituted ligand L1 and the monoamide functionalised pillar P2 with Zn2+, and exhibits the same pcu topology as the base structure, with paddlewheel SBUs connecting the diacids in two-dimensional sheets along the ab crystallographic plane and dipyridyl pillars linking the sheets along the c axis. In common with the parent, unfunctionalised species, the MOF shows two-fold interpenetration, but the introduction of the amide moiety into the pillar results in a change in orientation of the SBUs with respect to one another. The near rectangular arrangement of SBUs seen across the Zn2+-L1 sheets in the unfunctionalised analogue is distorted to a parallelogram arrangement in [Zn2(L1)2(P2)]n, with inter-SBU angles of 75.8° and 104.2°. The offset introduced (Fig. 2a) by the amide unit in P2 also results in distortions down the c axis, along which the pillars link the sheets, with inter-SBU angles of 74.6°, 79.7°, 100.3° and 105.4° deviating significantly from right angles. Each amide unit forms (Fig. 2b) a hydrogen bond to an ordered DMF solvent molecule, from the amido NH to the formamide CO, at a N⋯O distance of 2.79 Å, with flanking CH⋯O interactions from the β-aromatic CH units of each ring to the formamide oxygen atom (C⋯O = 3.12 Å, C⋯O = 3.38 Å) supporting the molecular recognition of the substrate. The carbonyl oxygen of the amide moiety in P2 is also involved in a CH⋯O interaction, with P2 linkers from individual nets forming reciprocal dimers (C⋯O = 3.35 Å) that ensure the close proximity of the two interpenetrated nets of pcu topology (Fig. 2c).
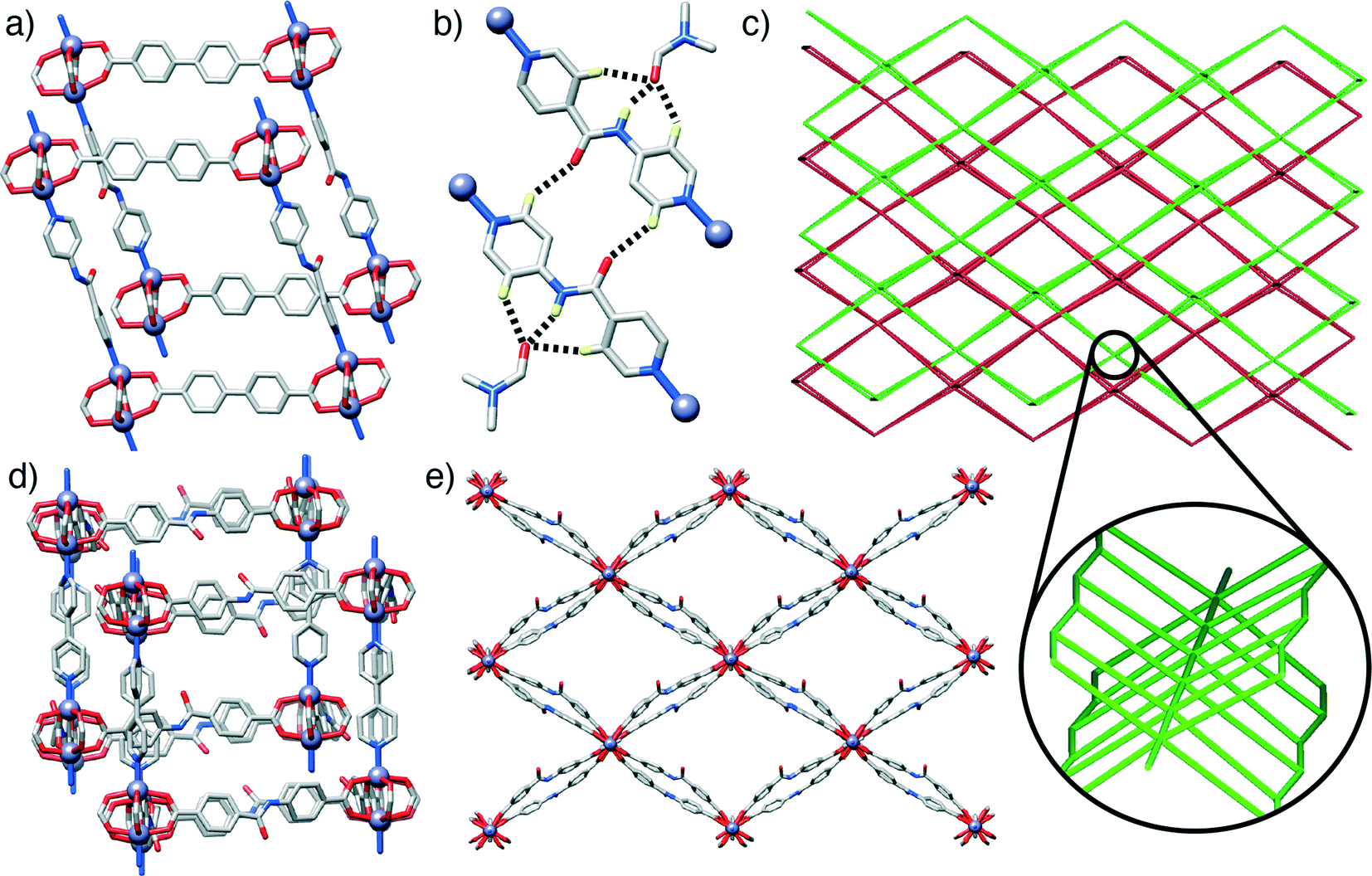 | ||
| Fig. 2 a) Portion of the solid-state structure of [Zn2(L1)2(P2)]n, showing the disposition of the SBUs induced by the amide pillar. b) The molecular recognition of a DMF solvate molecule by the amide unit of P2, which also forms a complementary H-bonded pair with a P2 unit of a separate net. c) The pcu topology present in both 1 and 2 with an inset showing the connection in the third dimension. d) Portion of the solid-state structure of 2, which has a more regular arrangement of SBUs compared to 1. e) Packing diagram of one of the two interpenetrated nets of [Zn2(L2)2(P1)]n, viewed down the crystallographic c axis, showing the alternating pattern of L2 linkers. | ||
Compound 2, [Zn2(L2)2(P1)]n, has an amide functionality incorporated in its acid linker, L2, rather than in the pillar, which is the unfunctionalised P1. A structure analogous to the previous examples again forms; a 2-fold interpenetrated pcu material (Fig. 2c), with Zn2+ paddlewheel SBUs linking the ligands. In a manner similar to 1, the arrangement of SBUs in the Zn2+-L2 plane is distorted from the parent structure, with inter-SBU angles of 70.1°, 70.3°, 109.1° and 110.4° resulting from the offset nature of amido-substituted L2. The unsubstituted pillar P1 links the sheet at angles close to 90°, however, in a manner similar to the parent structure (Fig. 2d). The sheets of acid ligands adopt an alternating arrangement (Fig. 2e), likely induced by a complementary pair of short CH⋯O contacts (C⋯O = 3.34 Å) between the amide oxygen of one molecule of L2 and an aromatic CH of another from the second, interpenetrating net. Half of the L2 units are involved in this interaction, while the other half form a bifurcated hydrogen bond to an ordered DMF guest, with the amido NH and one aromatic β-CH forming interactions with the formamide CO (N⋯O = 2.96 Å, C⋯O = 3.34 Å).
In contrast to the previous structures, compound 3, [Zn(L1)(P3)]n, obtained through the introduction of the urea-functionalised pillar P3 to the synthesis, results in an altogether different structure and topology. Instead of the dimeric paddlewheel SBU, a monomeric Zn2+ cation coordinates (Fig. 3a) two carboxylic groups of two unsubstituted L1 molecules and two N donors of two molecules of P3, forming a distorted octahedral coordination sphere that yields a distorted tetrahedral 4-connected node, with inter-SBU angles of 95.1°, 95.1°, 101.6°, 110.0°, 110.0° and 140.0° deviating significantly from the ideal 109.5°. Four identical diamondoid (dia) nets are interpenetrated (Fig. 3b), with DMF solvent molecules occupying significant rectangular channels (Fig. 3c) running down the crystallographic b axis, which may impart permanent porosity on the framework if successfully removed without collapse. Interestingly, the structure shows an important interaction between the interpenetrated nets (Fig. 3d), through a H-bond between a urea NH of each P3 and a carboxylic O of each L1 (N⋯O = 2.97 Å). This is also manifested in the distorted Zn–O bond lengths, with distances of 2.564 and 1.951 Å for the H-bonded and the non-interacting oxygens of the carboxylate, respectively.
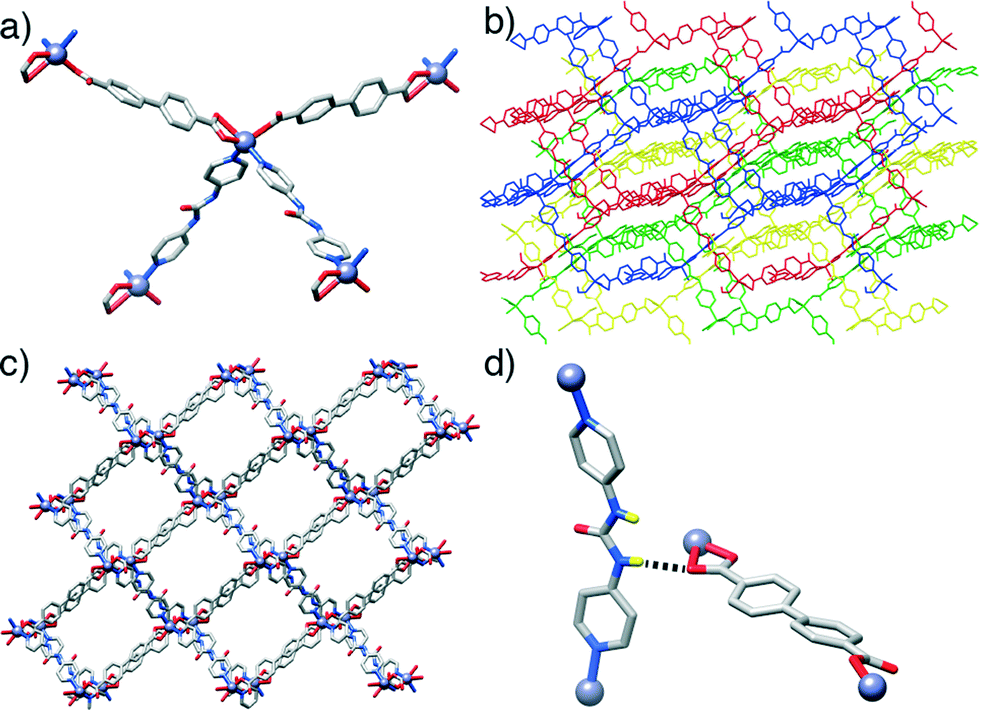 | ||
| Fig. 3 Representations of the crystal structure of [Zn(L1)(P3)]n. a) The four-connected Zn2+ node which connects the ligands. b) Packing structure showing the four-fold interpenetration of the material, with each net coloured differently. c) An alternative view of the packing structure to show the one dimensional channels that run down the crystallographic b axis. d) The hydrogen bond interaction between a urea proton of P3 and a carboxylic oxygen atom of L1 in an adjacent net. | ||
In common with 3, compound 4, [Zn(L2)(P3)]n, forms an interpenetrated dia topology, with a distorted octahedral Zn2+ coordination sphere consisting of two molecules each of L2 and P3. The amido-functionalised ligand L2 is disordered across two positions, with the offset of the amide moiety resulting in large thermal ellipsoids for the adjacent aromatic carbon atoms of the ligand. Compared to 3, the structure is also four-fold interpenetrated, with slightly smaller but still distinct one-dimensional channels running down the crystallographic a axis. Disorder in both the solvent and the amide unit make intermolecular interactions difficult to ascertain but, as in compound 4, a bifurcated interaction between the NH units of the urea pillar P3 and a carboxylic O atom from L2 in an adjacent net is observed (N⋯O = 2.76, N⋯O = 2.85 Å). Again, the H-bonding is reflected in the Zn–O bond distances, with values of 1.979 Å and 2.566 Å for the non-interacting and H-bonded oxygen atoms, respectively. Different nets also interact via π–π stacking between pyridino rings of adjacent molecules of P3, at a distance of 3.27 Å.
Some trends are noticeable from the structural data of these four compounds. The introduction of the urea-functionalised pillar P3 gives MOFs of a dramatically different dia topology compared to the pcu topology seen for both the unfunctionalised parent structure and the two amide-containing frameworks. Presumably, the bent nature of the urea moiety, which results in the non-linear (ca. 160°) disposition of the N-donors of the pyridyl units of P3, makes the dia topology more favourable. While the four compounds have structures comprising interpenetrating nets, those based on the longest pillar P3 lead to the formation of a more open framework and a higher degree of interpenetration, i.e. four-fold interpenetrated dia nets, classified as IIIa type. On the contrary, those composed of shorter pillars P1 and P2, give only two-fold interpenetrated pcu-nets, corresponding to class IIa.19 This is in agreement with the origin of interpenetration, since it can be ascribed to the presence of large free voids in a single network20 and it is well known that one effective way to increase the number of interpenetrating folds is to extend the length of the ligand sustaining the framework (Fig. 4).21
 | ||
| Fig. 4 a) Crystal packing in the solid-state structure of [Zn(L2)(P3)]n viewed down the crystallographic a axis to show one-dimensional pores. b) A bifurcated hydrogen bonding interaction in the crystal structure, between the urea unit of P3 and a carboxylic oxygen atom of a molecule of L2 from an adjacent net. c) The four-fold interpenetrated dia topology, with inset, adopted by both [Zn(L1)(P3)]n and [Zn(L2)(P3)]n. | ||
The introduction of functional groups into the frameworks results in H-bonds between the urea NH of each P3 and a carboxylic O of each L2 or L1, for compounds 3 and 4 respectively, of adjacent nets in the solid-state interpenetrated structures, possibly acting as structure-directing effects.22 Unmasking these functional groups to act as substrate binding sites would necessitate breaking of these interactions during activation. It should also be noted that choice of solvent can affect the structures formed by these functionalised ligands and pillars. A hydrothermal synthesis involving L1, P1, Zn2+ and base has been shown, by Rietveld refinement of powder X-ray diffraction data, to yield23 a non-porous coordination polymer, with five-fold interpenetrated dia topology, while a solvothermal synthesis using L2, P1 and Zn2+ in aqueous DMF gave24 a triply interpenetrated structure linked by an alternative paddlewheel SBU into an elongated pcu topology with overall formula [Zn(L2)(P1)]n. In this regard, the pcu topology has a higher degree of interconnectivity compared to the dia topology, and therefore higher metal–ligand interactions that in the absence of additional external factors may provide robustness to the framework as well as minimise the energy. Higher metal–ligand interactions and, consequently, higher node-connectivity are frequently associated with increasing synthesis temperature,25 which, in turn, favours the formation of thermodynamically more stable phases.26
In an attempt to rationalise the formation of the different topologies and to understand the effect of functional group incorporation on materials properties, we turned to molecular simulations in order to build hypothetical new structures corresponding to the synthesised materials with swapped linkers, i.e. allowing us to examine the arrangement of ligands and pillars in topologies in which they are not observed experimentally. That is, compound 1 was used as a parent structure of three new hypothetical ones using the ligand and pillar combinations of compounds 2–4, and this was repeated for each experimental structure. Therefore, from the initial 4 structures we built 12 new ones, named as [X-(L)(P)]*, where X is the mother structure, L and P correspond to the ligands and pillars used according to Fig. 1, and * denotes a computational structure. The strategy is similar to the methodology we have used previously for rht topology17 and alternative pillared MOFs.13e,f We start with the asymmetric unit of every mother structure, maintaining the metal cluster (i.e. Zn paddle wheel or Zn), the pcu or dia topology as well as the corresponding group symmetry, but we swap the organic linkers. After the asymmetric units are modified, the symmetry operations of the space group allow the construction of the new structures. The unit cells are then subject to energy minimisation and geometry optimisation based on molecular mechanics, which include van der Waals and electrostatic interactions, modifying the size of the unit cells and the atomic coordinates of the new structures.
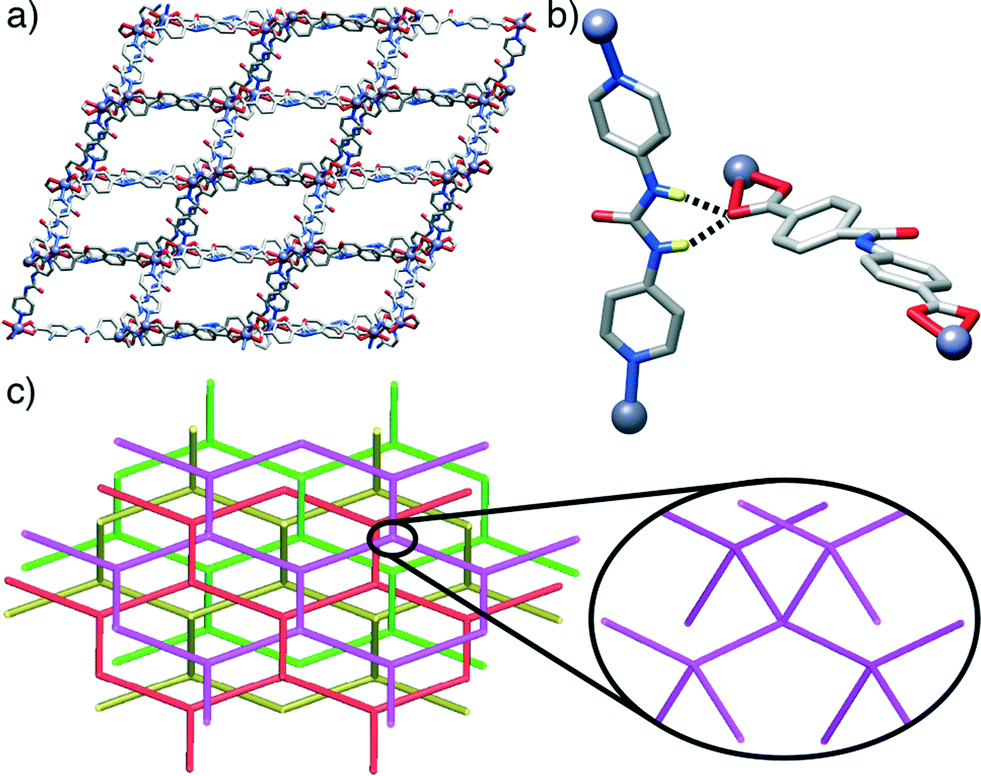
Interestingly, pcu topology hypothetical structures built with the L1 and L2 ligands and P3 pillar (i.e. those with the constituents of the dia topology structures 3 and 4) do not show any H-bonding between the urea moieties of each P3 and carboxylic units of either L1 or L2. This observation contrasts with the experimental dia compounds 3 and 4, which show these close interactions. Fig. 5 illustrates how, in the case of the hypothetical [1-(L1)(P3)]* structure, the urea NH groups from P3 point to the centre of the main cavities, precluding any possible interaction with L1. This observation reinforces our hypothesis about the importance of the H-bonds between adjacent nets present in compounds 3 and 4 in the formation and stabilisation of MOFs with dia topology instead of pcu. Indeed, it suggests that dia topology is effectively allowed and preferred when the urea moiety is incorporated into the pillar.
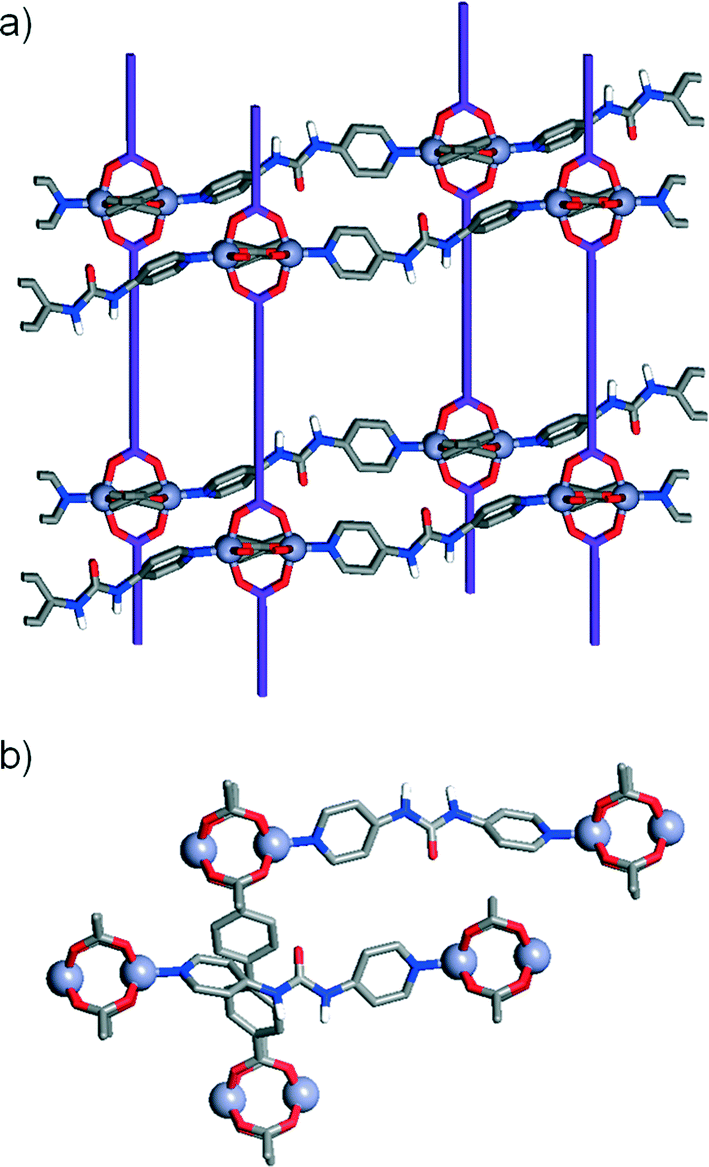 | ||
| Fig. 5 a) Crystal packing of and b) detail of the solid-state structure of [1-(L1)(P3)]*. Ligands L1 have been simplified for clarity and are represented in purple in (a). Note that the urea protons of P3 are pointing to the centre of the cavity and not to the carboxylic group of L1. | ||
The fact that compounds 3 and 4 show a higher degree of interpenetration is not unexpected, since the dia topology is the most preferred one for interpenetrating frameworks. This is because these arrays can be more easily assembled from tetrahedral coordination centres than from octahedral (in particular, most of the arrays with high interpenetration degree Z > 6 are diamondoid). The most abundant topologies are the most symmetrical ones because they provide the most isotropic medium during crystallization. For instance, the diamond net has the automorphism group isomorphic to the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group and may have an infinite number of distorted geometrical realizations (embeddings) of the space group symmetries corresponding to subgroups of Fdm. As it was demonstrated, any pair of nets can interpenetrate in some embedding, but in crystals there are physical reasons (like the condition of close packing), which essentially restrict possible combinations of interpenetrating nets.27 These observations support once more the importance of these interactions between interpenetrated structures and the possibility of the urea acting as a structure-directing agent.
m space group and may have an infinite number of distorted geometrical realizations (embeddings) of the space group symmetries corresponding to subgroups of Fdm. As it was demonstrated, any pair of nets can interpenetrate in some embedding, but in crystals there are physical reasons (like the condition of close packing), which essentially restrict possible combinations of interpenetrating nets.27 These observations support once more the importance of these interactions between interpenetrated structures and the possibility of the urea acting as a structure-directing agent.
To investigate the propensity for urea moieties to form noncovalent interactions with carboxylate units in the solid state, we examined existing structures deposited with the Cambridge Structural Database.28 Using a fragment-based search approach, 16 crystal structures, ranging from 1D coordination polymers to 3D MOFs, were identified, all of which containing both a carboxylate group coordinated to a metal and a N,N′-disubstituted urea function in an extended network. Of these 16 structures, 10 contained H-bonding interactions between urea NH units and carboxylate oxygen donors; eight involved interactions between individual ribbons of one dimensional coordination polymers29 and two between distinct two dimensional grids.29c Significant hydrogen bonding interactions were also present in the remaining structures, between different coordination polymer strands (three by urea–urea hydrogen bonded tapes,30 and one through hydrogen bonding with a terminal water ligand29c) as well as H-bonding to solvent (two examples with sterically constrained ligands12a,31) and one example of hydrogen bonding to nitrate counterions as well as to carboxylate units.29c In all but two of these cases, the urea groups of individual ribbons, sheets and nets form significant H-bonding interactions with adjacent ribbons, sheets and nets. The abundance of these noncovalent contacts in the solid-state structures of coordination polymers indicates they represent a significant structure directing influence, and may explain the preference for 3 and 4 to adopt the dia topology.
In order to compare the textural properties of the synthesised and hypothetical compounds, we first measured their geometrical surface area32 by rolling a 3.681 Å-diameter sphere, which corresponds to a nitrogen molecule, across the surface of the material. The pore size distributions (PSD) were calculated by recording the largest sphere that can fit in a random point within a structure without overlapping the van der Waals surface of the framework.33 In addition, we calculated the N2 adsorption isotherms at 77 K of the synthesised materials using grand canonical Monte Carlo (GCMC) simulations34 to predict their potential adsorption performance. Table S1† shows the geometrical surface area of all the materials. Fig. 6 shows the simulated N2 adsorption isotherms and the geometric PSD. Geometrical surface areas of all the synthesised compounds are very similar, with values ranging from 2000 and 2400 m2 g−1, and all four compounds present microporous cavities with diameters ranging from 6 to 8 Å. The simulated N2 isotherms each present a type I shape and show total pore volumes between 0.65 and 0.77 cm3 g−1. The textural properties (e.g. PSD, geometrical surface area and total pore volume) are directly related, with compound 2 showing the largest PSD, geometrical surface area and total pore volume, followed by 1 and 4. In addition, the N2 adsorption isotherm of compound 2 is displaced to higher relative pressures when compared with the other isotherms. The shape of the adsorption isotherms is related to the balance between the strength of the fluid–fluid and fluid–solid interactions, which in turn is a strong function of the length of the linker and therefore the pore size.35
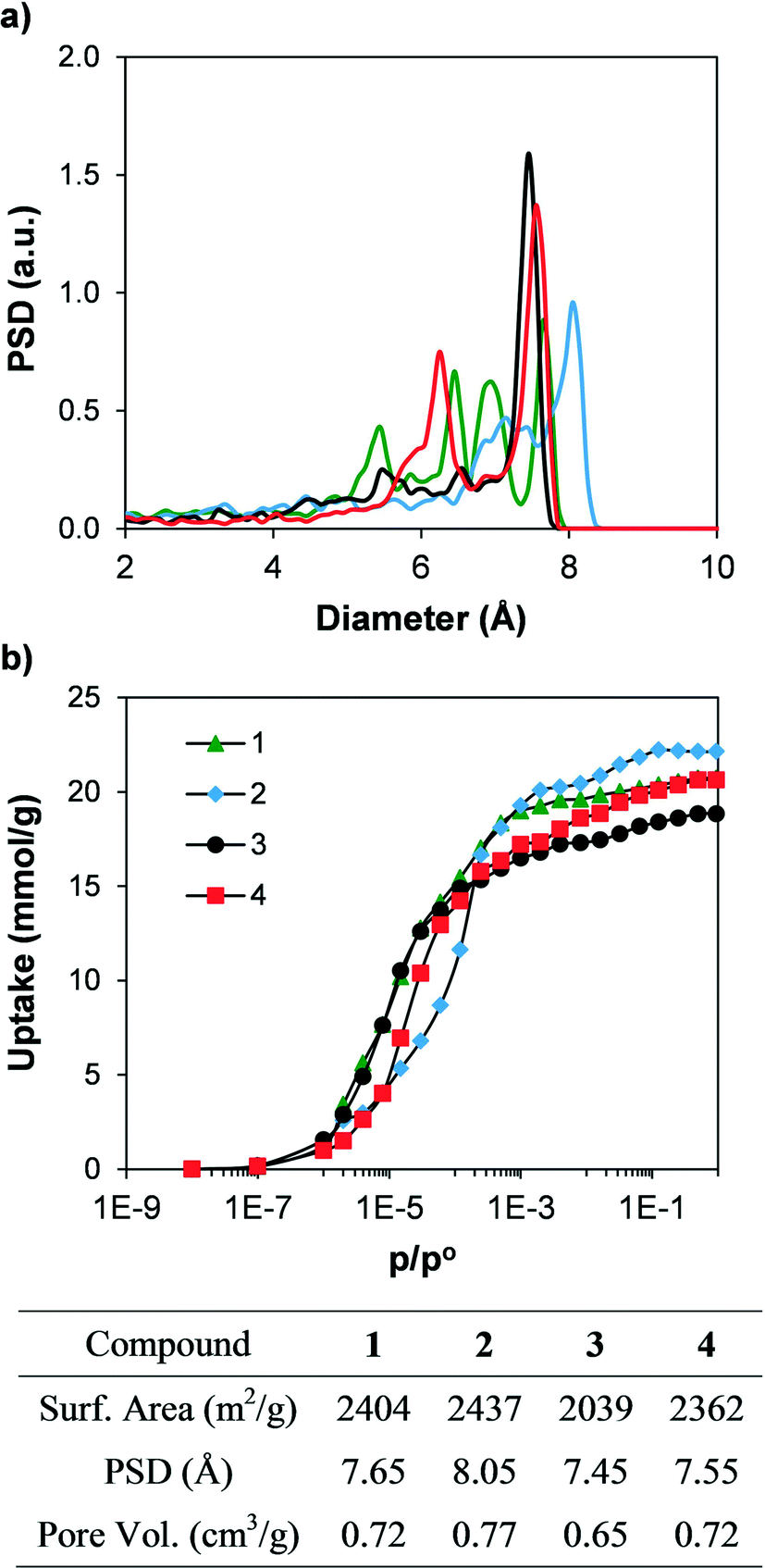 | ||
| Fig. 6 a) GCMC simulated adsorption isotherms of N2 at 77 K, b) geometric pore size distribution of the synthesised compounds. The table shows the geometrical surface area, pore size distribution (PSD) and total pore volume of the synthesised compounds. | ||
3. Conclusions
We have utilised a combination of crystallographic and computational analyses to investigate the systematic incorporation of amide and urea functional groups directly into metal–organic frameworks with pcu and dia topology. Introduction of amide units is tolerated within the pcu topology (compounds 1 and 2). However, integration of urea functional groups was not found to be possible under the synthetic conditions examined and led to the isolation of four-fold interpenetrated networks of dia topology with different compositions and SBUs (compounds 3 and 4). The comparison of hypothetical computationally created and experimental structures has shown that the presence of significant hydrogen bonding interactions between urea moieties of the pillars and carboxylate groups of the acidic linkers may contribute to the formation of the dia topology found in structures 3 and 4. Prediction of MOF topology and structure from a certain combination of ligands and metals remains difficult but possible,36 with control over variables such as solvent, temperature and concentration required. Our results demonstrate that noncovalent interactions between linkers may be significant structure-directing factors, which has implications for the integration of functional groups into MOFs to alter molecular recognition and catalytic properties.4. Experimental
All the frameworks described were prepared by dissolution of the ligand, pillar and [Zn(NO3)2]·6H2O in DMF, with crystallisation induced by heating the colourless solution in a sealed vial for a period of approximately 2–5 days. Detailed descriptions, alongside ligand syntheses and computational details, are given in the ESI.† CCDC 1011797–1011800 contain the supplementary crystallographic data for this paper.Acknowledgements
RSF and DFJ thank the Royal Society for receipt of University Research Fellowships. This research was funded in part by the EPSRC under grant EP/L004461/1 (RSF) and EPSRC IAA Partnership Development Award RG/75759 (DFJ). We thank the Cronin group at the University of Glasgow for technical support and equipment access.References
- (a) S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed; (b) S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490–2509 CrossRef CAS PubMed; (c) G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC; (d) J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC , and other articles in this themed issue; (e) H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed , and other articles in this themed issue.
- (a) R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef CAS PubMed; (b) L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC; (c) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC; (d) J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS PubMed; (e) M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed; (f) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- (a) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed; (b) B. Van de Voorde, B. Bueken, J. Denayer and D. De Vos, Chem. Soc. Rev., 2014, 43, 5766–5788 RSC.
- (a) D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed; (b) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; (c) L. Q. Ma, C. Abney and W. B. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC; (d) A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed; (e) J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS; (f) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- (a) P. Horcajada, C. Serre, M. Vallet-Regì, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 5974–5978 CrossRef CAS PubMed; (b) J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS PubMed; (c) I. Imaz, M. Rubio-Martinez, J. An, I. Sole-Font, N. L. Rosi and D. Maspoch, Chem. Commun., 2011, 47, 7287–7302 RSC; (d) P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed; (e) M. C. Bernini, D. Fairen-Jimenez, M. Pasinetti, A. J. Ramirez-Pastor and R. Q. Snurr, J. Mater. Chem. B, 2014, 2, 766–774 RSC.
- (a) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC; (b) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed; (c) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed; (d) D. Liu, K. Lu, C. Poon and W. Lin, Inorg. Chem., 2014, 53, 1916–1924 CrossRef CAS PubMed; (e) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- (a) Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC; (b) A. D. Burrows, CrystEngComm, 2011, 13, 3623–3642 RSC; (c) S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- (a) Y. Zou, S. Hong, M. Park, H. Chun and M. S. Lah, Chem. Commun., 2007, 5182–5184 RSC; (b) R. Vaidhyanathan, S. S. Iremonger, K. W. Dawson and G. K. H. Shimizu, Chem. Commun., 2009, 5230–5232 RSC; (c) S. Couck, J. F. M. Denayer, G. V. Baron, T. Rémy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS PubMed; (d) J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 132, 38–39 CrossRef PubMed; (e) A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS PubMed; (f) R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed; (g) T. Panda, P. Pachfule, Y. Chen, J. Jiang and R. Banerjee, Chem. Commun., 2011, 47, 2011–2013 RSC; (h) X. Wang, H. Li and X.-J. Hou, J. Phys. Chem. C, 2012, 116, 19814–19821 CrossRef CAS; (i) A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gándara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863–8866 CrossRef CAS PubMed.
- (a) J. J. Gassensmith, H. Furukawa, R. A. Smaldone, R. S. Forgan, Y. Y. Botros, O. M. Yaghi and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 15312–15315 CrossRef CAS PubMed; (b) R. S. Forgan, R. A. Smaldone, J. J. Gassensmith, H. Furukawa, D. B. Cordes, Q. Li, C. E. Wilmer, Y. Y. Botros, R. Q. Snurr, A. M. Z. Slawin and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 406–417 CrossRef CAS PubMed; (c) D. Wu, J. J. Gassensmith, D. Gouvêa, S. Ushakov, J. F. Stoddart and A. Navrotsky, J. Am. Chem. Soc., 2013, 135, 6790–6793 CrossRef CAS PubMed.
- (a) B. Zheng, J. Bai, J. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 748–751 CrossRef CAS PubMed; (b) C. Hou, Q. Liu, T.-A. Okamura, P. Wang and W.-Y. Sun, CrystEngComm, 2012, 14, 8569–8576 RSC.
- (a) J. Duan, Z. Yang, J. Bai, B. Zheng, Y. Li and S. Li, Chem. Commun., 2012, 48, 3058–3060 RSC; (b) B. Zheng, Z. Yang, J. Bai, Y. Li and S. Li, Chem. Commun., 2012, 48, 7025–7027 RSC; (c) S. Xiong, Y. He, R. Krishna, B. Chen and Z. Wang, Cryst. Growth Des., 2013, 13, 2670–2674 CrossRef CAS; (d) B. Zheng, H. Liu, Z. Wang, X. Yu, P. Yi and J. Bai, CrystEngComm, 2013, 15, 3517–3520 RSC; (e) B. Liu, D.-S. Li, L. Hou, G.-P. Yang, Y.-Y. Wang and Q.-Z. Shi, Dalton Trans., 2013, 42, 9822–9825 RSC; (f) J. Duan, M. Higuchi, M. L. Foo, S. Horike, K. P. Rao and S. Kitagawa, Inorg. Chem., 2013, 52, 8244–8249 CrossRef CAS PubMed; (g) C.-H. Lee, H.-Y. Huang, Y.-H. Liu, T.-T. Luo, G.-H. Lee, S.-M. Peng, J.-C. Jiang, I. Chao and K.-L. Lu, Inorg. Chem., 2013, 52, 3962–3968 CrossRef CAS PubMed; (h) L. Du, S. Yang, L. Xu, H. Min and B. Zheng, CrystEngComm, 2014, 16, 5520–5523 RSC; (i) N. H. Alsmail, M. Suyetin, Y. Yan, R. Cabot, C. P. Krap, J. Lü, T. L. Easun, E. Bichoutskaia, W. Lewis, A. J. Blake and M. Schröder, Chem. – Eur. J., 2014, 20, 7317–7324 CrossRef CAS PubMed.
- (a) J. M. Roberts, B. M. Fini, A. A. Sarjeant, O. K. Farha, J. T. Hupp and K. A. Scheidt, J. Am. Chem. Soc., 2012, 134, 3334–3337 CrossRef CAS PubMed; (b) P. W. Siu, Z. J. Brown, O. K. Farha, J. T. Hupp and K. A. Scheidt, Chem. Commun., 2013, 49, 10920–10922 RSC.
- (a) K. Seki, Chem. Commun., 2001, 1496–1497 RSC; (b) K. Seki and W. Mori, J. Phys. Chem. B, 2002, 106, 1380–1385 CrossRef CAS; (c) D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033–5036 CrossRef CAS PubMed; (d) B.-Q. Ma, K. L. Mulfort and J. T. Hupp, Inorg. Chem., 2005, 44, 4912–4914 CrossRef CAS PubMed; (e) W. Bury, D. Fairen-Jimenez, M. B. Lalonde, R. Q. Snurr, O. K. Farha and J. T. Hupp, Chem. Mater., 2013, 25, 739–744 CrossRef CAS; (f) O. Karagiaridi, W. Bury, D. Fairen-Jimenez, C. E. Wilmer, A. A. Sarjeant, J. T. Hupp and O. K. Farha, Inorg. Chem., 2014, 53, 10432–10436 CrossRef CAS PubMed.
- P. V. Dau, M. Kim, S. J. Garibay, F. H. L. Münch, C. E. Moore and S. M. Cohen, Inorg. Chem., 2012, 51, 5671–5676 CrossRef CAS PubMed.
- (a) I. A. Baburin, S. Leoni and G. Seifert, J. Phys. Chem. B, 2008, 112, 9437–9443 CrossRef CAS PubMed; (b) D. W. Lewis, A. R. Ruiz-Salvador, A. Gomez, L. M. Rodriguez-Albelo, F.-X. Coudert, B. Slater, A. K. Cheetham and C. Mellot-Draznieks, CrystEngComm, 2009, 11, 2272–2276 RSC; (c) I. A. Baburin and S. Leoni, J. Mater. Chem., 2012, 22, 10152–10154 RSC; (d) R. Galvelis, B. Slater, A. K. Cheetham and C. Mellot-Draznieks, CrystEngComm, 2012, 14, 374–378 RSC.
- (a) A. M. Walker, B. Civalleri, B. Slater, C. Mellot-Draznieks, F. Corà, C. M. Zicovich-Wilson, G. Román-Pérez, J. M. Soler and J. D. Gale, Angew. Chem., Int. Ed., 2010, 49, 7501–7503 CrossRef CAS PubMed; (b) T. D. Bennett, J.-C. Tan, S. A. Moggach, R. Galvelis, C. Mellot-Draznieks, B. A. Reisner, A. Thirumurugan, D. R. Allan and A. K. Cheetham, Chem. – Eur. J., 2010, 16, 10684–10690 CrossRef CAS PubMed; (c) D. Fairen-Jimenez, R. Galvelis, A. Torrisi, A. D. Gellan, M. T. Wharmby, P. A. Wright, C. Mellot-Draznieks and T. Düren, Dalton Trans., 2012, 41, 10752–10762 RSC.
- D. Fairen-Jimenez, Y. J. Colon, O. K. Farha, Y.-S. Bae, J. T. Hupp and R. Q. Snurr, Chem. Commun., 2012, 48, 10496–10498 RSC.
- In all experiments involving L3, we were unable to isolate any materials containing both L3 and a pillar. In each case, a MOF comprising only Zn2+ and L3, with a structure analogous to a known material containing Zn2+ and L2 (NJU-Bai-2) was observed. See: J. Duan, J. Bai, B. Zheng, Y. Li and W. Ren, Chem. Commun., 2011, 47, 2556–2558 RSC.
- V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2004, 6, 378–395 RSC.
- (a) J. Yang, J.-F. Ma, S. R. Batten and Z.-M. Su, Chem. Commun., 2008, 2233–2235 RSC; (b) H. Wu, H.-Y. Liu, Y.-Y. Liu, J. Yang, B. Liu and J.-F. Ma, Chem. Commun., 2011, 47, 1818–1820 RSC.
- S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460–1494 CrossRef.
- Inter-ligand hydrogen bonding, similar to peptide β-sheets, has been observed in coordination polymers of P2 and Ag+. See: K. Uemura, Y. Kumamoto and S. Kitagawa, Chem. – Eur. J., 2008, 14, 9565–9576 CrossRef CAS PubMed.
- K. O. Kongshaug and H. Fjellvåg, J. Solid State Chem., 2003, 175, 182–187 CrossRef CAS.
- X.-M. Lian, W. Zhao and X.-L. Zhao, J. Solid State Chem., 2013, 200, 265–270 CrossRef CAS PubMed.
- P. M. Forster, A. R. Burbank, C. Livage, G. Ferey and A. K. Cheetham, Chem. Commun., 2004, 368–369 RSC.
- (a) A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 4780–4795 RSC; (b) M. C. Bernini, V. A. de la Peña O'Shea, M. Iglesias, N. Snejko, E. Gutierrez-Puebla, E. V. Brusau, G. Narda and M. A. Monge, Inorg. Chem., 2010, 49, 5063–5071 CrossRef CAS PubMed; (c) A. E. Platero-Prats, V. A. de la Peña-O'Shea, D. M. Proserpio, N. Snejko, E. Gutierrez-Puebla and A. Monge, J. Am. Chem. Soc., 2012, 134, 4762–4771 CrossRef CAS PubMed.
- E. V. Alexandrov, V. A. Blatov, A. V. Kochetkov and D. M. Proserpio, CrystEngComm, 2011, 13, 3947–3958 RSC.
- CSD Version 5.35 (November 2013) was searched using CCDC ConQuest Version 1.16. Full search parameters are available in the ESI.†.
- (a) Y. Hao, B. Wu, S. Li, C. Jia, X. Huanga and X.-J. Yangab, CrystEngComm, 2011, 13, 215–222 RSC; (b) B.-C. Tzeng, Y.-C. Huang, B.-S. Chen, W.-M. Wu, S.-Y. Lee, G.-H. Lee and S.-M. Peng, Inorg. Chem., 2007, 46, 186–195 CrossRef CAS PubMed; (c) S. Banerjee, D. P. Kumar, S. Bandyopadhay, N. N. Adarsh and P. Dastidar, Cryst. Growth Des., 2012, 12, 5546–5554 CrossRef CAS; (d) D. K. Kumar, A. Das and P. Dastidar, CrystEngComm, 2006, 8, 805–814 RSC; (e) P. Díaz, J. Benet-Buchholz, R. Vilar and A. J. P. White, Inorg. Chem., 2006, 45, 1617–1626 CrossRef PubMed.
- (a) M. Stapf, T. Bohle, W. Seichter, F. O. R. L. Mertens and E. Weber, Z. Naturforsch., B: J. Chem. Sci., 2012, 67, 1166–1172 CrossRef CAS; (b) A. Karmakar and J. B. Baruah, Inorg. Chem. Commun., 2009, 12, 140–144 CrossRef CAS PubMed.
- N. Qureshi, D. S. Yufit, J. A. K. Howard and J. W. Steed, Dalton Trans., 2009, 5708–5714 RSC.
- T. Düren, F. Millange, G. Férey, K. S. Walton and R. Q. Snurr, J. Phys. Chem. C, 2007, 111, 15350–15356 Search PubMed.
- L. D. Gelb and K. E. Gubbins, Langmuir, 1999, 15, 305–308 CrossRef CAS.
- (a) D. Frenkel and B. Smit, Understanding Molecular Simulations: From Algorithms to Applications, Academic Press, San Diego, 2nd edn, 2002 Search PubMed; (b) D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Düren, J. Am. Chem. Soc., 2011, 133, 8900–8902 CrossRef CAS PubMed.
- D. Fairen-Jimenez, N. A. Seaton and T. Düren, Langmuir, 2010, 26, 14694–14699 CrossRef CAS PubMed.
- V. Guillerm, Ł. J. Weseliński, Y. Belmabkhout, A. J. Cairns, V. D'Elia, Ł. Wojtas, K. Adil and M. Eddaoudi, Nat. Chem., 2014, 6, 673–680 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1011797–1011800. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce01379d |
| This journal is © The Royal Society of Chemistry 2015 |