
Fluorinated azobenzenes with highly strained geometries for halogen bond-driven self-assembly in the solid state†
Oleksandr S.
Bushuyev
,
Davin
Tan
,
Christopher J.
Barrett
* and
Tomislav
Friščić
*
Department of Chemistry and FRQNT Centre for Self-Assembled Chemical Structures, McGill University, 801 Sherbrooke St. W., H3A 0B8 Montreal, Quebec, Canada. E-mail: tomislav.friscic@mcgill.ca; christopher.barrett@mcgill.ca; Tel: +1 514 398 3959
First published on 22nd September 2014
Abstract
Attempted cocrystallisation of brominated and iodinated octafluoroazobenzene derivatives with morpholine led to the exhaustive replacement of fluorine substituents that are in ortho-positions to the azobenzene with sterically demanding morpholine groups. The resulting molecules exhibit a highly unusual strained conformation of the azobenzene unit, in which the terminal phenyl rings adopt a mutually nearly completely orthogonal orientation. Substitution of ortho-fluorine groups with N-morpholine fragments provides molecules with active halogen bond donor and acceptor sites that guide the molecular self-assembly in the solid state towards the formation of polymeric halogen-bonded chains.
Introduction
Azobenzene (azo) compounds are a family of molecules that is ubiquitous in the design of photo-switchable materials, i.e. materials whose properties can be changed upon irradiation with light.1 The photo-switchable behaviour of azobenzene-based materials is based on a change of molecular geometry upon light-induced reversible cis–trans azo isomerisation.2 Owing to the large stereochemical change involved in the isomerisation, incorporation of azo molecules within polymers,3 liquid crystals4 and even crystalline metal–organic frameworks,5 has been utilized to create solid materials displaying the photomechanical effect, i.e. the direct conversion of light into mechanical energy.6 Very recently, however, it was demonstrated that crystalline solid azobenzenes can exhibit reversible7 or irreversible8 photo-mechanical behaviour, inspiring interest and activity in structural characterization of azobenzene solids. In that context, we have recently demonstrated irreversible solid-state cis–trans isomerisation in crystals of cis-4,4′-di(bromo)perfluoroazobenzene (cis-1) and its iodo analogue (cis-2).8 The solid-state isomerisation allowed the controllable and permanent modification in shape of the crystalline solids through irreversible photomechanical transformation. More recently, Saccone and co-workers have demonstrated the synthesis of two-component cocrystals using trans-2 as the halogen bond donor in combination with nitrogen-based halogen bond9 acceptors,10 and our group has demonstrated irreversible photomechanical bending induced by visible light irradiation of halogen-bonded cocrystals of cis-1 and cis-2.11We now describe the crystal structures of three substituted derivatives of azobenzenes 1 and 2 that were serendipitously obtained as a result of attempted halogen bond-driven cocrystallisation with morpholine. In particular, recrystallisation of either cis-1 or trans-1 from liquid morpholine at room temperature produced, instead of the desired halogen-bonded cocrystals with morpholine, compounds 3 and 4 in which three or four of the fluorine substituents in ortho-position to the azo group are substituted by N-morpholine residues (Scheme 1), respectively. Similarly, attempted recrystallisation of trans-2 or cis-2 from morpholine gave compound 5 in which all fluorine groups ortho to the azo moiety are replaced by N-morpholine substituents.
 | ||
| Scheme 1 The preparation of herein investigated compounds 3–5trans-5. | ||
The herein described crystal structures of 3–5 are remarkable in terms of molecular structure, as the bulkiness of juxtapositioned N-morpholine substituents imposes a highly unusual stereochemistry of the azo moiety, as well as in terms of crystal packing due to the presence of strong halogen bond donor and acceptor sites on each molecule.
Experimental
The trans- and cis-forms of the precursors 1 and 2 were synthesised using previously described procedures.8 Compounds 3–5 were prepared by dissolving 50 mg cis- or trans-1 or -2 in morpholine at room temperature. Reaction mixture immediately turned red and colour rapidly became deeper, reaching almost black within 15 minutes. The formation of fluoride ions in the reaction was confirmed by recording an 19F NMR spectrum of the reaction mixture involving cis-1, which revealed the appearance of a signal at 146 ppm, consistent with the shift of a fluoride ion in a separately prepared sample of morpholinium fluoride (see ESI†). After 12 hours, the solvent was removed on a rotary evaporator and samples were purified using an automated CombiFlash column equipped with a 4 g silica cartridge and a mixture of hexanes and ethyl acetate as the mobile phase. The identity of products 3–5 was confirmed by X-ray analysis, mass spectrometry (MS), 1H NMR, 19F NMR (see ESI†) and elemental analysis. 3: 4 mg (5.6%) 1H NMR (300 MHz, CDCl3): δ 3.59–3.55 (comp, 8H), δ 3.15–3.06 (comp, 8H), MS m/z 686.02 C24H24Br2F5N5O3 (M + 1), decomposes without melting at 171 °C; calculated for 3: N (10.22%) C (42.06%) H (3.53%), determined for 3: N (10.08%) C (42.33%) H (3.42%); 4: 41 mg (53% yield), 1H NMR (300 MHz, CDCl3): δ 3.51–3.49 (t, J = 3.0 Hz, 8H) v δ 3.11–3.09 (t, J = 3.0 Hz, 8H), MS m/z 753.08 C28H32Br2F4N6O4 (M + 1), decomposes without melting at 175 °C; calculated for 4·morpholine: N (11.68%) C (45.78%) H (4.92%), determined for 4·morpholine: N (11.55%) C (46.18%) H (5.03%); 5: 43 mg (58%), 1H NMR (300 MHz, CDCl3): δ 3.51–3.49 (t, J = 3.0 Hz, 8H), δ 3.08 (app. s, 8H), MS m/z 847.05 C28H32I2F4N6O4 (M + 1), decomposes without melting at 175 °C; calculated for 5·morpholine: N (10.50%) C (41.17%) H (3.43%), determined for 5·morpholine: N (10.00%) C (41.35%) H (4.34%). Fourier-transform infrared (FTIR) spectra of all three compounds are very similar and are given in the ESI.†Single crystals were prepared by recrystallisation of ca. 10 mg of the corresponding precursors 1 or 2 from excess morpholine. Using this methodology, single crystals of 3 were always obtained in small amounts along with the crystals of the main product 4·morpholine.
Single crystal X-ray diffraction experiments (Table 1) were performed on a Bruker D8 single crystal diffractometer at 100 K or 150 K. Crystal structure refinement was conducted using SHELXL-2014/3 implemented within the WinGX 2013.3 integrated program system.12,13 Solution UV–vis measurements were performed on a Cary 300 Bio spectrometer using THF, DCM and cyclohexane as solvents. In all solvents we found that solutions of 4 and 5 exhibit nearly identical spectra, including a shoulder at 500 nm of coalescing π–π* and n–π* transitions (see ESI, Fig. S1†). Cambridge Structural Database (CSD) searches were performed using the CSD version 5.35, November 2013 update.
| Compound | 3 | 4·morpholine | 5·morpholine |
|---|---|---|---|
| a The crystal diffracted poorly past 2θ = 45°. | |||
| Formula | C24H24Br2F5N5O3 | C32H41Br2F4N7O5 | C32H41F4I2N7O5 |
| CCDC code | 1007719 | 1007720 | 1007721 |
| Crystal system | Monoclinic | Orthorhombic | Orthorhombic |
| Space group | P21/c | Pna21 | Pna21 |
| a/Å | 14.052(3) | 19.4138(15) | 19.6801(13) |
| b/Å | 10.029(3) | 17.3697(14) | 17.4108(11) |
| c/Å | 18.994(5) | 10.4512(8) | 10.6952(7) |
| β/° | 101.069(3) | 90 | 90 |
| V/Å3 | 2627.0(11) | 3524.3(5) | 3664.7(4) |
| Z | 4 | 4 | 4 |
| T/K | 150(2) | 150(2) | 100(2) |
| Flack parameter | — | 0.024(6) | 0.003(11) |
| ρ calc/g cm−3 | 1.73 | 1.58 | 1.69 |
| No. data | 4628 | 8163 | 8076 |
| No. data with I ≥ 2σI | 1910 | 5730 | 6845 |
| No. parameters | 352 | 452 | 451 |
| R int | 0.187a | 0.086 | 0.066 |
| F(000) | 1368 | 1712 | 1856 |
| R (all data) | 0.199 | 0.091 | 0.054 |
| R (I ≥ 2σI) | 0.069 | 0.049 | 0.039 |
| wR2 (all data) | 0.180 | 0.101 | 0.074 |
| wR2 (I ≥ 2σI) | 0.148 | 0.088 | 0.069 |
| S | 0.925 | 1.023 | 1.015 |
Density Functional Theory (DFT) calculations were performed using the Gaussian09 package of programs.14 Geometrical optimisations were carried out using the popular Becke's three parameter functional15 with the nonlocal Lee–Yang–Parr correlation functional16 (B3LYP) theory. LANL2DZ basis set including double-ζ valence basis set with the Hay and Wadt effective core potential17 (ECP) was used for the heavy Br and I atoms, while the 6-31+G(d,p) Pople basis set18 was used for the rest of the atoms.
Results and discussion
Molecular and crystal structures of 3 and 4·morpholine
Crystallisation of 1 from morpholine gave rise to two distinct types of crystals: a small number of carmine red plates and much more abundant burgundy red needles. Crystal structure analysis (Table 1) revealed that the plates consist of molecules (3) in which three out of four fluorine substituents ortho to the azo moiety of trans-1 have been replaced by nitrogen-bound morpholine units (Fig. 1a). | ||
| Fig. 1 (a) Thermal ellipsoid plot of a single molecule of 3 as found in the crystal structure, thermal ellipsoids are shown at 25% probability level. Overlay of the solid-state conformations of 3 (green) to that of trans-1 (red) viewed: along (b) and perpendicular (c) to the molecular plane of trans-1. For clarity all phenyl ring substituents except bromine atoms have been removed in the overlay figures (b) and (c); (d) the assembly of two molecules of 3 in the solid state, based on a pair of Br⋯O interactions. | ||
Due to steric crowding around the azo group, the molecules of 3 adopt a conformation that is very different from that found in the parent trans-1. Specifically, the trans-azo group, defined by the two nitrogen atoms and the two adjacent carbon atoms, is no longer planar but exhibits a significant torsional angle (τ) of 15(1)°. Moreover, the phenyl ring substituents that are coplanar in the crystals of trans-1 starting material are now found to be almost completely perpendicular, with an angle (π) of 88.8(3)° between the best planes drawn through the carbon atoms of each phenyl ring (Fig. 1b, c). In the crystal, the molecules of 3 are associated by relatively long19 Br⋯O interactions of 3.272(8) Å into discrete halogen-bonded dimers. Consistent with their length, these halogen-bonding interactions show a significant deviation from linearity,20 expressed by the C–Br⋯O angle of 168.8(3)°. No specific interactions are observed between the dimers, except a pair of long C–H⋯O21 interactions (C⋯O distance 3.59(1) Å).
Crystallographic analysis of burgundy red needles that are the major product of recrystallising trans- or cis-1 from morpholine revealed a morpholine solvate of an even more sterically hindered molecule (4), in which all four fluorine substituents ortho to the azo moiety have been replaced by N-morpholine residues (Fig. 2a).
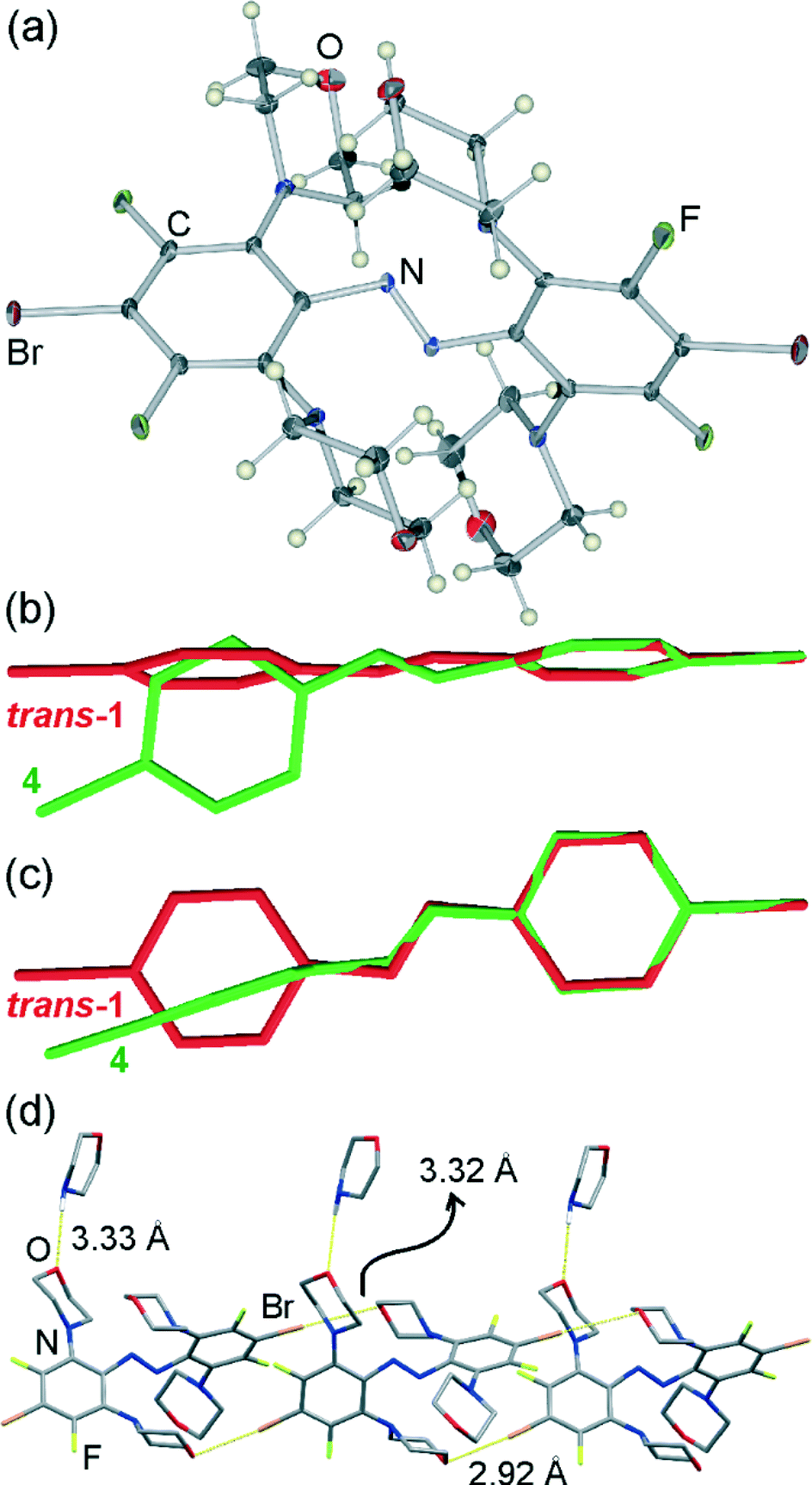 | ||
| Fig. 2 Thermal ellipsoid plot of a single molecule of 4 as found in the crystal structure, with thermal elliposids shown at 25% probability level. Overlay of the solid-state conformations of one molecule of 4 (green) to that of trans-1 (red) viewed: (b) along and (c) perpendicular to the molecular plane of trans-1. For clarity all phenyl ring substituents except the bromine atoms have been removed in the overlay figures (b) and (c). (d) A single halogen-bonded chain of 4 decorated by hydrogen-bonded morpholine molecules in the crystal structure of 4·morpholine, with hydrogen atoms of all hydrocarbon groups omitted for clarity. | ||
Similar to 3, the trans-azo group in 4 is no longer planar but exhibits a torsional angle τ = 14.6(5)°, and the phenyl groups are mutually perpendicular with the angle π = 87.5(2)° between the planes drawn through carbon atoms of each ring (Fig. 2b, c). In a crystal of 4·morpholine, molecules of 4 are associated into one-dimensional halogen-bonded polymers held together by Br⋯O interactions and propagating in the crystallographic (011) direction (Fig. 2d). Each molecule of 4 is involved in two shorter and two longer halogen bonds, simultaneously acting as a two-fold halogen bond donor and a two-fold halogen bond acceptor. Two of the halogen bonds to each molecule are significantly shorter (Br⋯O distance 2.923(5) Å) than those observed in the crystal structure of 3, while the other two are of comparable length (Br⋯O distance 3.324(5) Å).
Consistent with previous studies, the shorter halogen bonds exhibit a higher degree of linearity (C–Br⋯O angle of 176.0(2)°) than the longer ones (C–Br⋯O angle of 165.6(2)°). The additional molecules of morpholine in the crystal structure are associated with the halogen-bonded chains of 4 by way of long N–H⋯O hydrogen bonds (N⋯O distance 3.33(1) Å, Fig. 2d).
Crystal and molecular structure of 5·morpholine
Molecular structures of 3 and 4 are remarkable not only because of their highly unusual stereochemistry, but also in the context of supramolecular chemistry. These serendipitously obtained molecules contain both a strong halogen bond donor site, in the form of an electron-deficient bromine atom attached on a fluorinated benzene ring, as well as a potential halogen bond acceptor site in the form of the ether oxygen22 of the N-morpholine substituents (Fig. 3a). | ||
| Fig. 3 (a) Electrostatic potential plot of a single molecule of 4, revealing well-defined areas of negative and positive potential located on the oxygen and bromine atoms, respectively; (b) a thermal ellipsoid plot of a single molecule of 5 as found in the crystal structure of 5·morpholine, with thermal elliposids shown at 25% probability level; (c) electrostatic potential plot of a single molecule of 5; (d) overlay of the molecular geometries of 4 (blue) and 5 (red) as found in solid-state solvates with morpholine. For clarity, hydrogen atoms are omitted. (e) A single halogen-bonded chain of 5 decorated by hydrogen-bonded morpholine molecules in the crystal structure of 5·morpholine, with hydrogen atoms of all hydrocarbon groups omitted for clarity. | ||
Whereas the intense exploration of halogen bonding in the solid state has strongly focused on the formation of cocrystals between halogen bond donors and acceptors, there has been much less exploration of molecular building blocks designed to form self-assembled halogen-bonded structures in single-component crystals.23 The facile transformation of trans- or cis-1 into 3 or 4 upon recrystallisation from morpholine offers a simple means to create such mixed donor and acceptor building blocks. As the first step in that direction, we attempted the analogous replacement of fluorine atoms in 2, the iodinated analogue of 1. Indeed, recrystallisation of either trans-2 or cis-2 from morpholine gave crystals whose unit cell parameters were found to be almost identical to those of 4·morpholine. Crystal structure determination revealed that the crystal consists of molecules of 5, an iodine-based analogue of 4, and morpholine solvent in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio. The crystal of 5·morpholine is isostructural to that of 4·morpholine, with 5 adopting a molecular conformation almost identical to that observed for 4 (Fig. 3b–d). Like in 4·morpholine, the molecules of 5 in the structure of 5·morpholine are associated into infinite halogen-bonded chains, propagating in the crystallographic (011) direction and held together by I⋯O halogen bonds (Fig. 3e).
:1 stoichiometric ratio. The crystal of 5·morpholine is isostructural to that of 4·morpholine, with 5 adopting a molecular conformation almost identical to that observed for 4 (Fig. 3b–d). Like in 4·morpholine, the molecules of 5 in the structure of 5·morpholine are associated into infinite halogen-bonded chains, propagating in the crystallographic (011) direction and held together by I⋯O halogen bonds (Fig. 3e).
In the chain, molecules of 5 act as a two-fold halogen bond donors and two-fold acceptors. Each molecule forms one pair of shorter (I⋯O distance 2.923(5) Å) and one pair of longer (3.140(5) Å) I⋯O halogen bonding interactions. The shorter ones are highly linear (C–I⋯O angle 175.1(2)°) and comparable to the analogous interactions in 4·morpholine (Fig. 3e). The other set of I⋯O halogen bonds are considerably shorter and more linear (C–I⋯O angle of 172.2(2)°) than their Br⋯O counterparts in 4·morpholine, consistent with the greater strength and linearity of halogen bonds involving iodine donors24,25 compared to those based on bromine. The molecules of morpholine in the crystal structure of 5·morpholine are associated with halogen-bonded chains of 5 through long N–H⋯O interactions whose length (3.25(1) Å) and geometry resemble those found in 4·morpholine (Fig. 3e). The similarity of crystal structures of 4·morpholine and 5·morpholine is consistent with the previously described isostructurality of halogen-bonded structures based on iodine and bromine.19b,25 Another view of the crystal structures of 4·morpholine and 5·morpholine is as lattice inclusion compounds involving halogen-bonded chains of 5 as the host and morpholine molecules as guests. In such a view, the lattice host is formed from halogen-bonded chains which stack in the crystallographic bc-plane to form layers. The assembly of neighbouring layers into a stacked log pattern creates cavities that host morpholine guests (Fig. 4a, b).
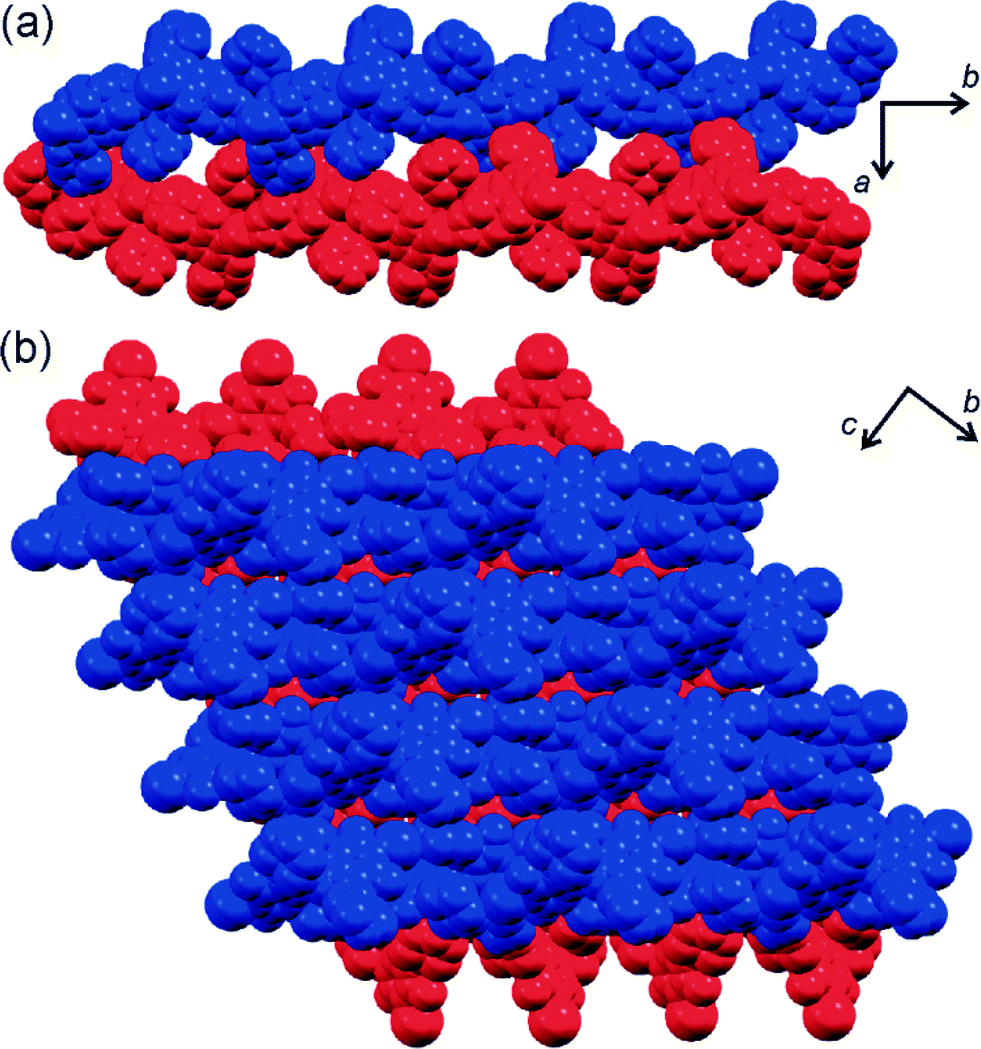 | ||
| Fig. 4 (a) Formation of cavities by the assembly of halogen-bonded chains in 5·morpholine, with morpholine molecules occupying the cavities omitted and (b) the “log stack” assembly of layers of parallel halogen-bonded chains of 5. For clarity, different layers are coloured red and blue. | ||
Database analysis
Analysis of the Cambridge Structural Database (CSD) reveals that the geometry of the azo moiety in 3 and 4 is among the most distorted ones reported so far in trans-azobenzenes. The database search focused on the distribution of the torsion angle around the azo moiety (τ, Fig. 5a) and π, the angle between the planes of the two azobenzene phenyl substituents. Most reported trans-azobenzene structures exhibit highly planar molecular conformations, reflected by a π angle of no more than 50°, with the torsion angle τ deviating no more than ±10° from 180°. A limited number of structures exhibit a perpendicular orientation of phenyl substituents which, upon individual inspection, appears to be associated with the presence of sterically highly demanding substituents in ortho-positions to the azo group (e.g. in BRBUAB, τ = 3.2°, π = 87.3° or HNIABZ21 τ = 1.5°, π = 85.2°).26 The distortion of the torsion angle τ is encountered much more rarely and appears to be associated with strained structures, e.g. with the azobenzene group being a part of a cyclophane ring (e.g. in JEMLIX, τ = 15.3°, π = 84.0°)27 or highly sterically encumbered systems, such as 2,2′,4,4′,6,6′-hexakis(tert-butyl)azobenzene (CSD KEBYAT, τ = 10.1°, π = 83.0°).28 The combination of unusual τ and π angles places the conformations of 3 and 4 among the most extreme distortions of the trans-azobenzene moiety reported in a crystalline solid (Fig. 5b).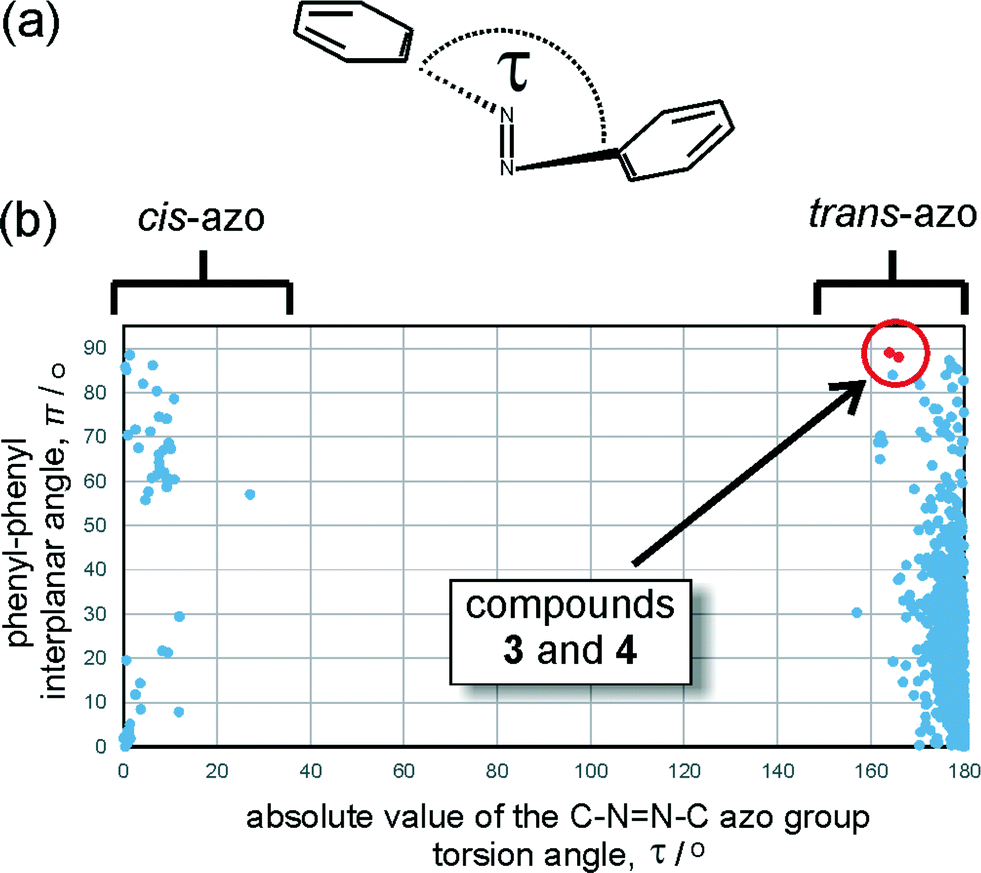 | ||
Fig. 5 (a) Schematic representation of the C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C torsion angle (τ) around the azo moiety, used in the CSD search. (b) Scatterplot of the absolute value of τ against the phenyl–phenyl interplanar angle (π), based on azobenzene structures deposited in the CSD. Molecular geometries of 3 and 4 are highlighted in red. The highly populated region at τ = 180° corresponds to trans-azobenzene structures, while the poorly populated region around τ = 0° corresponds to structures of cis-azobenzenes. N–C torsion angle (τ) around the azo moiety, used in the CSD search. (b) Scatterplot of the absolute value of τ against the phenyl–phenyl interplanar angle (π), based on azobenzene structures deposited in the CSD. Molecular geometries of 3 and 4 are highlighted in red. The highly populated region at τ = 180° corresponds to trans-azobenzene structures, while the poorly populated region around τ = 0° corresponds to structures of cis-azobenzenes. | ||
Computational studies
In order to evaluate the degree of distortion of the azobenzene moiety imposed by introducing the four bulky substituents in compound 4, we have compared the calculated molecular energies for azobenzene, perfluoroazobenzene and compound 1 in three different conformations: the idealised planar arrangement found in crystal structures of all three compounds,8,29–31 the minimum energy gas-phase conformation calculated at the B3LYP level and the orthogonal conformation corresponding to that of the azobenzene skeleton in the crystal structure of 4.Gas-phase energy minimisation confirmed the planar conformation as the most stable one for azobenzene. However, calculations on perfluoroazobenzene and 1 revealed that these molecules can access a twisted conformation ca. 2 kcal mol−1 lower than the planar conformations found in crystal structures. Comparison of the molecular energies calculated for all three conformations indicated that the orthogonal conformation adopted by molecules of 4 is up to 40 kcal mol−1 higher than the energy of the lowest energy conformation (Table 2). The orthogonal geometries were first obtained from the crystal structures of compound 4·morpholine by replacing the ortho morpholine moieties with fluorine atoms and subsequently freezing the core Ph–NN–Ph atoms while the other peripheral atoms were allowed to relax. Although the constraints required to impose this particular geometry imply that the calculated energies should be taken with caution, the obtained values clearly indicate a high degree of strain in the azobenzene skeleton of 4.
| Planar | Gas-phase minimum | Orthogonal | |
|---|---|---|---|
| Azobenzene | 0.0 | — | +31.5 |
| Perfluoroazobenzene | 0.0 | −1.9 | +38.3 |
| 1 | 0.0 | −2.0 | +39.7 |
It is surprising that the azobenzene skeleton can be twisted into a high-energy conformation via a simple and mild room-temperature procedure. Presumably, compounds 3–5 result from an electrophillic substitution of fluorine atoms ortho to an azo group, resulting in the formation of a new carbon–nitrogen bond and morpholinium fluoride byproduct. This pathway is confirmed by the 19F NMR spectrum of the reaction mixture involving cis-1, which revealed the formation of a signal at 137.5 ppm, consistent with that for a separately prepared sample of morpholinium fluoride (139 ppm, see ESI†). We computationally confirmed the likelihood of such a reaction by evaluating the free energy change for the transformation of compound 1 into 4 in the gas phase (Fig. 6). Calculated at the B3LYP level, the free energy change for this reaction is ca. −8 kcal mol−1. The calculations also revealed the LUMO levels of 1 are largely localised on azo nitrogen atoms, as well as the carbon atoms in ortho- and para-positions to the azo group (Fig. 6). Such LUMO distribution is consistent with the observed facile nucleophilic substitution of fluorine atoms ortho to the azo moiety. The calculations revealed a similar organisation of the LUMO levels for perfluoroazobenzene, indicating that the observed reactivity is not associated with the presence of the heavy halogen substituent in the para-position. A similar result was obtained by the Hecht group32 in their study of the electronic structure of trans-perfluoroazobenzene, supporting the validity of our results. The distribution of LUMO levels in compound 1 is not significantly different between the idealised planar conformation and the calculated lowest-energy conformation, indicating that the observed nucleophilic substitution reactions are not significantly affected by molecular conformation.33
 | ||
| Fig. 6 (top) Computationally evaluated gas-phase transformation of 1 into 4 and (bottom) plot of the LUMO level for a molecule of 1, calculated at the B3LYP level. | ||
Conclusions
In summary, we have recognised a simple room-temperature reaction which allows extensive substitution of all four sites ortho to the azo group in perhalogenated azobenzenes. The resulting azobenzenes are highly sterically hindered and exhibit two halogen bond donor and three or four halogen bond acceptor sites. Such distribution of substituents allows for the formation of self-assembled halogen-bonded chain structures in the solid state. Whereas the exploration of molecules capable of self-assembling by way of strong halogen bonds is still in its infancy, the herein presented molecules illustrate an opportunity to expand such studies towards potentially photo-dynamic systems involving the azo chromophore. The ability to readily generate new potentially functional systems capable of halogen bonding is, we believe, one of the most important requirements for the further development of halogen bonding as a concept relevant to a wide range of research areas, including functional materials science, pharmaceuticals and biomolecular studies.9,34 In contrast to conventional methods33 for making substituted perfluorobenzene halogen bond donors, which focus on modifying the substituent para to the halogen bond donor atom, the herein observed reactivity leads to the modification of substituents in meta-positions to the halogen bond donor. We are now exploring the synthetic scope of this facile method for derivatising fluorinated azobenzenes.Acknowledgements
We acknowledge Prof. D. S. Bohle for aid in obtaining single crystal X-ray diffraction data and M. J. Landry for help with 19F NMR experiments. O.S.B acknowledges financial support from a Vanier Canada Graduate Scholarship, D.T. acknowledges the financial support from NSERC CREATE program in Green Chemistry and C.J.B. a Fulbright Canada Fellowship. All authors are grateful for support from the NSERC Discovery Grant program, the Canada Foundation for Innovation Leader's Opportunity Fund, and the FRQNT Nouveaux Chercheurs program.Notes and references
- (a) C.-C. Ko and V. Wing-Wah Yam, in Supramolecular Chemistry, John Wiley & Sons, Ltd, 2012 Search PubMed; (b) C. J. Barrett, J.-I. Mamiya, K. G. Yager and T. Ikeda, Soft Matter, 2007, 3, 1249–1261 RSC.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- (a) T. J. White, N. V. Tabiryan, S. V. Serak, U. A. Hrozhyk, V. P. Tondiglia, H. Koerner, R. A. Vaia and T. J. Bunning, Soft Matter, 2008, 4, 1796 RSC; (b) D. H. Wang, K. M. Lee, Z. Yu, H. Koerner, R. A. Vaia, T. J. White and L.-S. Tan, Macromolecules, 2011, 44, 3840 CrossRef CAS; (c) A. Priimagi, G. Cavallo, A. Forni, M. Gorynsztejn-Leben, M. Kaivola, P. Metrangolo, R. Milani, A. Shishido, T. Pilati, G. Resnati and G. Terraneo, Adv. Mater., 2012, 22, 2572–2579 CAS.
- (a) H. F. Yu and T. Ikeda, Adv. Mater., 2011, 23, 2149 CrossRef CAS PubMed; (b) K. Min Lee, B. M. Lynch, P. Luchette and T. J. White, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 876 CrossRef CAS; (c) A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686 CrossRef CAS PubMed; (d) A. Priimagi, M. Saccone, G. Cavallo, A. Shishido, T. Pilati, P. Metrangolo and G. Resnati, Adv. Mater., 2012, 24, OP345–OP352 CAS.
- R. Lyndon, K. Konstas, B. P. Ladewig, P. D. Southon, P. C. J. Kepert and M. R. Hill, Angew. Chem., Int. Ed., 2013, 52, 3695 CrossRef CAS PubMed.
- M. Yamada, M. Kondo, J. I. Mamiya, Y. L. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986 CrossRef CAS PubMed.
- (a) H. Koshima, N. Ojima and H. Uchimoto, J. Am. Chem. Soc., 2009, 131, 6890 CrossRef CAS PubMed; (b) H. Koshima and N. Ojima, Dyes Pigm., 2012, 92, 798 CrossRef CAS; (c) O. S. Bushuyev, T. A. Singleton and C. J. Barrett, Adv. Mater., 2013, 25, 1796 CrossRef CAS PubMed; (d) N. K. Nath, L. Pejov, S. M. Nichols, C. Hu, N. I. Saleh, B. Kahr and P. Naumov, J. Am. Chem. Soc., 2014, 136, 2757 CrossRef CAS PubMed; (e) N. K. Nath, M. K. Panda, S. C. Sahoo and P. Naumov, CrystEngComm, 2014, 16, 1850–1858 RSC.
- O. S. Bushuyev, A. Tomberg, T. Friščić and C. J. Barrett, J. Am. Chem. Soc., 2013, 135, 12556 CrossRef CAS PubMed.
- (a) P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed; (b) G. R. Desiraju, P. S. Lo, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS; (c) J.-Y. Le Questel, C. Laurence and J. Graton, CrystEngComm, 2013, 15, 3212–3221 RSC.
- M. Saccone, G. Terraneo, T. Pilati, G. Cavallo, A. Priimagi, P. Metrangolo and G. Resnati, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 149 CAS.
- O. S. Bushuyev, T. C. Corkery, C. J. Barrett and T. Friščić, Chem. Sci., 2014, 5, 3158–3164 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Acta Crystallogr., Sect. B: Struct. Sci., 1988, 37, 785 CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 720 CrossRef.
- (a) S. K. Nayak, G. Terraneo, A. Forni, P. Metrangolo and G. Resnati, CrystEngComm, 2012, 14, 4259–4261 RSC; (b) D. Cinčić, T. Friščić and W. Jones, Chem. – Eur. J., 2008, 14, 747–753 CrossRef PubMed.
- (a) A. C. Legon, Phys. Chem. Chem. Phys., 2010, 12, 7736–7747 RSC; (b) M. Saccone, G. Cavallo, P. Metrangolo, A. Pace, I. Pibiri, T. Pilati, G. Resnati and P. Metrangolo, CrystEngComm, 2013, 15, 3102 RSC.
- For recent studies of the interplay of halogen bonding and C–H⋯O interactions in the solid state, see: (a) S. Y. Oh, C. W. Nickels, F. Garcia, W. Jones and T. Friščić, CrystEngComm, 2012, 14, 6110–6114 RSC; (b) R. H. Jones, K. S. Knight, W. G. Marshall, S. J. Coles, P. N. Horton and M. B. Pitak, CrystEngComm, 2013, 15, 8572–8577 RSC.
- For selected studies involving halogen bonding to oxygen acceptors, see: (a) K. S. Eccles, R. E. Morrison, S. P. Stokes, G. E. O'Mahony, J. A. Hayes, D. M. Kelly, N. M. O'Boyle, L. Fábián, H. A. Moynihan, A. R. Maguire and S. E. Lawrence, Cryst. Growth Des., 2012, 12, 2969–2977 CrossRef CAS; (b) L. Russo, S. Biella, M. Lahtinen, R. Liantonio, P. Metrangolo, G. Resnati and K. Rissanen, CrystEngComm, 2007, 9, 341–344 RSC; (c) A. Forni, P. Metrangolo, T. Pilati and G. Resnati, Cryst. Growth Des., 2004, 4, 291–295 CrossRef CAS; (d) C. B. Aakeröy, S. Panikkattu, P. D. Chopade and J. Desper, CrystEngComm, 2013, 15, 3125–3136 RSC.
- (a) J. D. Virdo, Y. H. Kawar, A. J. Lough and T. P. Bender, CrystEngComm, 2013, 15, 3187–3199 RSC; (b) S. M. Walter, M. G. Sarwar, M. G. Chudzinski, E. Herdtweck, A. J. Lough, S. M. Huber and M. S. Taylor, CrystEngComm, 2013, 15, 3097–3101 RSC; (c) R. W. Troff, T. Mäkela, F. Topić, A. Valkonen, K. Raatikainen and K. Rissanen, Eur. J. Org. Chem., 2013, 1617–1637 CrossRef CAS; (d) S. Guieu, J. Rocha and A. M. S. Silva, J. Mol. Struct., 2013, 1035, 1–5 CrossRef CAS; (e) E. Cariati, G. Cavallo, A. Forni, G. Leem, P. Metrangolo, F. Meyer, T. Pilati, G. Resnati, S. Righetto, G. Terraneo and E. Tordin, Cryst. Growth Des., 2011, 11, 5642–5648 CrossRef CAS; (f) J. Martí-Rujas, L. Colombo, J. Lü, A. Dey, G. Terraneo, P. Metrangolo, T. Pilati and G. Resnati, Chem. Commun., 2012, 48, 8207–8209 RSC; (g) T. Shirman, D. Freeman, Y. D. Posner, I. Feldman, A. Facchetti and M. E. van der Boom, J. Am. Chem. Soc., 2008, 130, 8162–8163 CrossRef CAS PubMed.
- (a) M. Kolář, J. Hostaš and P. Hobza, Phys. Chem. Chem. Phys., 2014, 16, 9987 RSC; (b) B. Pinter, N. Nagels, W. A. Herrebout and F. de Proft, Chem. – Eur. J., 2013, 19, 519–530 CrossRef CAS PubMed; (c) C. B. Aakeroy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem. – Eur. J., 2013, 19, 16240–16247 CrossRef PubMed.
- (a) D. Cinčić, T. Friščić and W. Jones, New J. Chem., 2008, 32, 1776–1781 RSC; (b) D. Cinčić, T. Friščić and W. Jones, Chem. Mater., 2008, 20, 6623–6624 CrossRef.
- (a) E. J. Gabe, Y. Wang and Y. Le Page, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 980–981 CrossRef; (b) E. J. Greber and B. Morosin, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 310–317 CrossRef; (c) M. A. Rodriguez, C. F. Campana, A. D. Rae, E. Graeber and B. Morosin, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, o127–o130 Search PubMed.
- (a) N. Tamaoki and M. Wada, J. Am. Chem. Soc., 2006, 128, 6284–6285 CrossRef CAS PubMed; (b) Y. Norikane, R. Katoh and N. Tamaoki, Chem. Commun., 2008, 1898 RSC.
- M. I. Lipschutz and T. D. Tilley, Chem. Commun., 2012, 48, 7146–7148 RSC.
- (a) J. Harada and K. Ogawa, J. Am. Chem. Soc., 2004, 126, 3539–3544 CrossRef CAS PubMed; (b) J. Harada, K. Ogawa and S. Tomoda, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 662–672 Search PubMed; (c) J. A. Bouwstra, A. Schouten and J. Kroon, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 1121–1123 CrossRef.
- K. Chinnakali and H.-K. Fun, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 615–616 CrossRef.
- For examples of computational studies of trans- and cis-azobenzenes, see: (a) D. L. Silva, N. A. Murugan, J. Kongsted, Z. Rinkevicius, S. Canuto and H. Ågren, J. Phys. Chem. B, 2012, 116, 8169–8181 CrossRef CAS PubMed; (b) H. Karabiyik, H. Petek, N. O. İskeleli and C. Albayrak, Struct. Chem., 2009, 20, 903–910 CrossRef CAS; (c) T. Tsuji, H. Takashima, H. Takeuchi, T. Egawa and S. Konaka, J. Phys. Chem. A, 2001, 105, 9347–9353 CrossRef CAS; (d) C. Cojocaru, A. Airinei and N. Fifere, SpringerPlus, 2013, 2, 586 CrossRef PubMed; (e) R. J. Maurer and K. Reuter, J. Chem. Phys., 2011, 135, 224303 CrossRef PubMed; (f) M. Dubecký, R. Derian, L. Mitas and I. Štich, J. Chem. Phys., 2010, 133, 244301 CrossRef PubMed; (g) T. Schultz, J. Quenneville, B. Levine, A. Toniolo, T. J. Martínez, S. Lochbrunner, M. Schmitt, J. P. Schaffer, M. Z. Zgierski and A. Stolow, J. Am. Chem. Soc., 2003, 125, 8098–8099 CrossRef CAS PubMed; (h) P. Duchstein, C. Neiss, A. Görling and D. Zahn, J. Mol. Model., 2012, 18, 2479–2482 CrossRef CAS PubMed.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- For similar fluorine substitution reactions applied towards molecules designed for halogen bonding, see: (a) P. Metrangolo, F. Meyer, T. Pilati and G. Resnati, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o211 CAS; (b) P. Metrangolo, F. Meyer, T. Pilati, D. M. Proserpio and G. Resnati, Cryst. Growth Des., 2008, 8, 654–659 CrossRef CAS; (c) A. Abate, S. Biella, G. Cavallo, F. Meyer, H. Neukirch, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, J. Fluorine Chem., 2009, 130, 1171–1177 CrossRef CAS; (d) G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, G. Terraneo and M. Ursini, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o579–o580 CAS; (e) T. Caronna, R. Liantonio, T. A. Logothetis, P. Metrangolo, T. Pilati and G. Resnati, J. Am. Chem. Soc., 2004, 126, 4500–4501 CrossRef CAS PubMed; (f) C. B. Aakeröy, A. Rajbanshi, P. Metrangolo, G. Resnati, M. F. Parisi, J. Desper and T. Pilati, CrystEngComm, 2012, 14, 6366–6368 RSC.
- (a) A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed; (b) M. R. Scholfield, C. M. Vander Zanden, M. Carter and P. S. Ho, Protein Sci., 2013, 22, 139–152 CrossRef CAS PubMed; (c) M. Baldrighi, G. Cavallo, M. R. Chierotti, R. Gobetto, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Mol. Pharmaceutics, 2013, 10, 1760–1772 CrossRef CAS PubMed; (d) A. Valkonen, M. Chukhlieb, J. Moilanen, H. M. Tuononen and K. Rissanen, Cryst. Growth Des., 2013, 13, 4769–4775 CrossRef CAS; (e) C. Präsang, L. J. McAllister, A. C. Whitwood and D. W. Bruce, CrystEngComm, 2013, 15, 8947 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, UV-vis spectra of 4 and 5 in tetrahydrofuran solution, FTIR spectra, results of thermal and mass spectroscopy and selected 19F spectra. Cartesian coordinates for calculated molecular structures and crystallographic data for 3, 4·morpholine and 5·morpholine in CIF format (respective CCDC deposition numbers 1007719–1007721). See DOI: 10.1039/c4ce01216j |
| This journal is © The Royal Society of Chemistry 2015 |