
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric fluorinative dearomatization of tryptamine derivatives†
Xiao-Wei
Liang
,
Chuan
Liu
,
Wei
Zhang
and
Shu-Li
You
 *
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Science, 345 Lingling Lu, Shanghai 200032, China. E-mail: slyou@sioc.ac.cn
First published on 24th April 2017
Abstract
An asymmetric fluorinative dearomatization reaction of tryptamine derivatives was developed by using a chiral anion phase transfer catalyst (PTC) system, and the preliminary results of the reaction mechanistic study were achieved. This method is characterized by a simple operation, facile introduction of a fluorine atom in a highly enantioselective manner and construction of two contiguous quaternary stereogenic centers.
Fluorination reactions have witnessed tremendous development due to the extreme importance of fluorine-containing molecules in medicinal chemistry and materials science.1 Although catalytic asymmetric fluorination reactions have gained rapid growth accompanied by the emergence of catalytic systems,2 developing highly efficient stereoselective fluorination reactions is still in great demand. Catalytic asymmetric dearomatization (CADA) reactions have recently attracted enormous research interest due to their powerful ability to construct complex molecules from readily available aromatic compounds.3 Although CADA reactions through fluorination are undoubtedly attractive in the synthesis of chiral fluorine-containing compounds and highly desirable, successful reports are very limited in number. In 2011, Gouverneur and co-workers reported an elegant enantioselective dearomatization of indole derivatives via cascade fluorocyclization. In general, moderate to high levels of enantioselective control are obtained under catalytic conditions (Scheme 1).4 Almost at the same time, Toste and co-workers reported enantioselective electrophilic fluorination utilizing a chiral anion phase transfer catalyst.5 The examples of dearomatization of benzothiophenes reported therein elucidated the potential of CADA reactions via fluorination. Later, the same group further advanced the asymmetric fluorinative dearomatization reaction using phenols.6 Due to the great demand for complex chiral fluorinated molecules and our continuous interest in CADA reactions, herein, we report an asymmetric fluorinative dearomatization reaction of tryptamine derivatives, providing fluorine-containing pyrroloindolines bearing two contiguous quaternary stereogenic centers. Ma and co-workers had reported the chiral anion PTC strategy in bromination of tryptamine derivatives. Under the same catalytic conditions, the fluorination of 1a gave almost racemic results.7
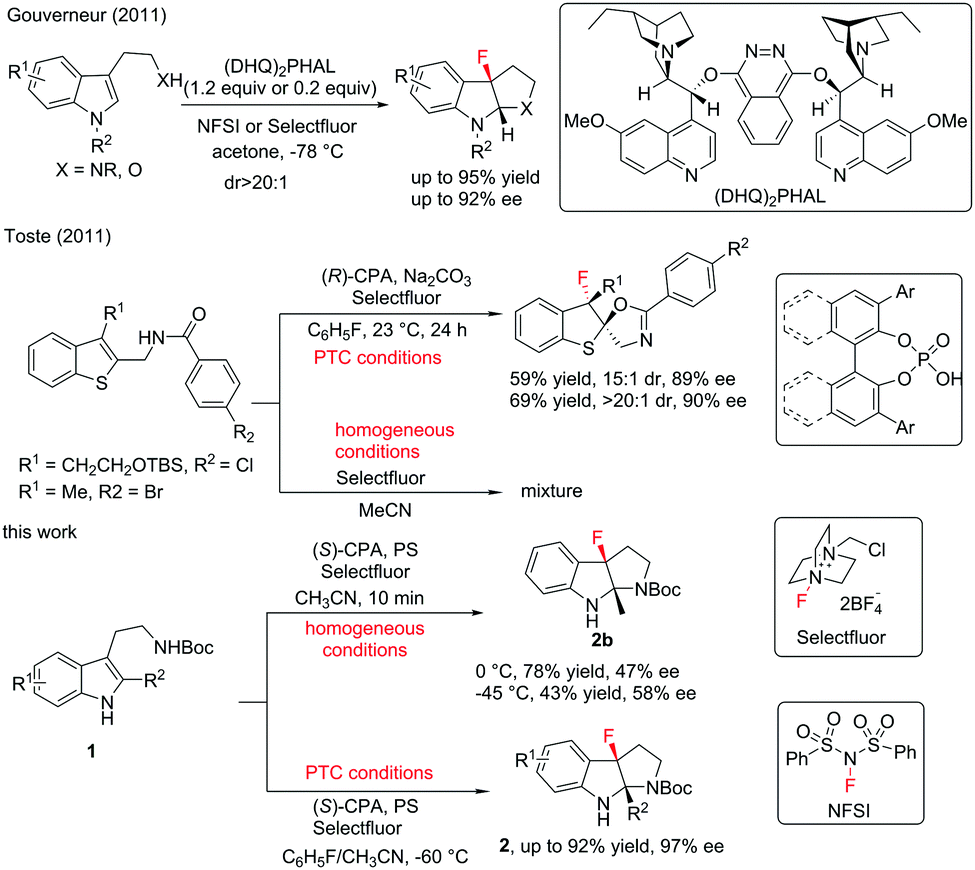 | ||
| Scheme 1 Enantioselective fluorinative dearomatization reactions of tryptamine derivatives. | ||
We initiated our studies by examining the reaction of the tryptamine derivative 1a with Selectfluor ((1-(chloromethyl)-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane)) (1.1 equiv.), in the presence of (S)-TRIP ((S)-3,3′-bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogen phosphate) (10 mol%) and sodium carbonate (1.1 equiv.) in fluorobenzene (Table 1, entry 1). The fluorocyclization product 2a was given in 48% yield and 6% ee.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entrya | CPA | 1 | Yieldb (%) | eec (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reactions were performed with 1 (0.2 mmol), Selectfluor (0.22 mmol), Na2CO3 (0.22 mmol), CPA (0.02 mmol) in C6H5F (4 mL) at rt. b Isolated yield. c Determined by HPLC analysis. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | C1 | 1a | 48 | 6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | C2 | 1a | 70 | 4 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | C3 | 1a | 52 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | C4 | 1a | 58 | 13 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | C5 | 1a | 48 | 3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | C6 | 1a | 67 | 37 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | C7 | 1a | 56 | 22 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | C6 | 1b | 48 | 55 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
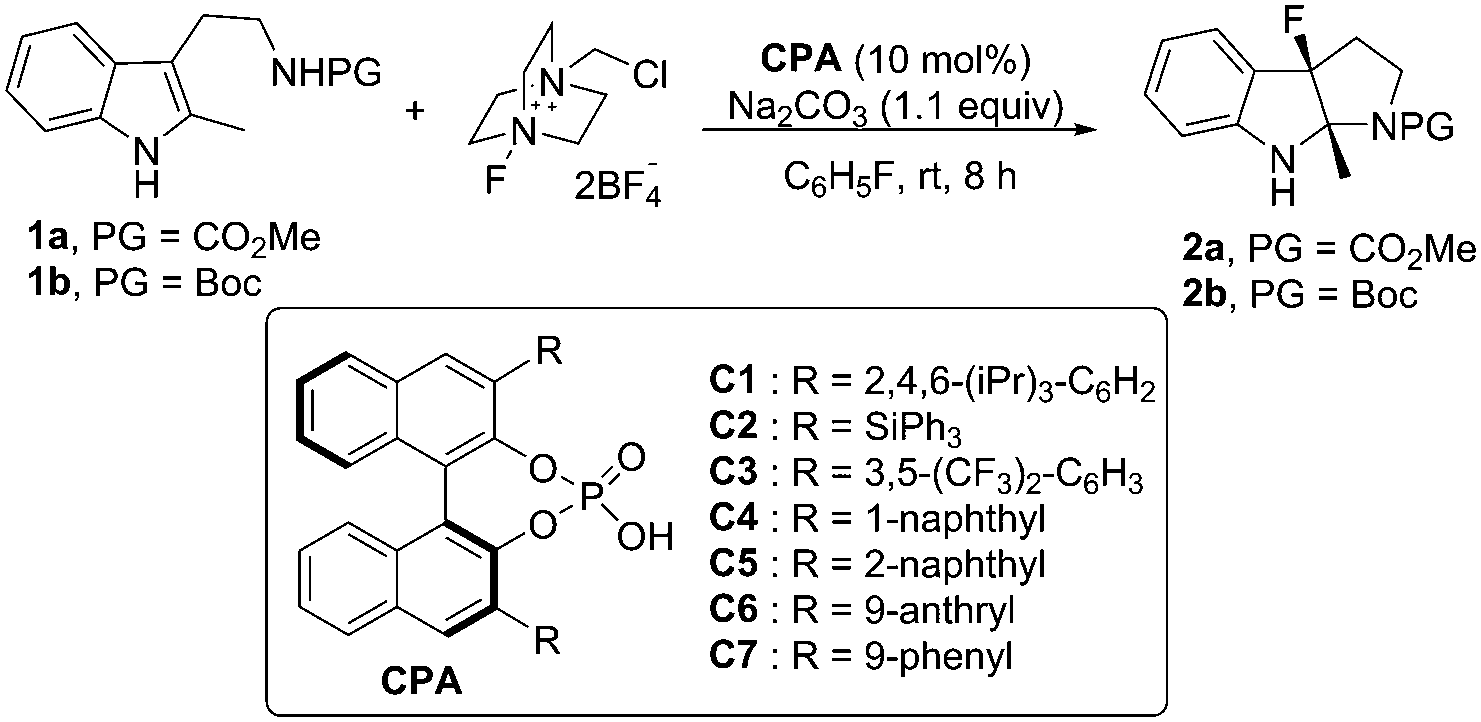
Encouraged by these results, various chiral phosphoric acids (CPAs) (10 mol%) were evaluated.8 Although all reactions proceeded to afford the desired fluorinated 6H-pyrroloindole92a, only moderate yields could be achieved and the enantioselective control was not satisfactory (Table 1, entries 1 to 7). Among all the CPAs surveyed, C6 was the optimal one to give 2a in 67% yield with 37% ee (Table 1, entry 6). To our delight, when the protecting group of tryptamine derivative 1 was switched from CO2Me (1a) to Boc (1b), product 2b was obtained in 55% ee (Table 1, entry 7).
With 1b as the model substrate, several commonly used non-polar solvents were first screened, and fluorobenzene proved to be the optimal one (48% yield, 55% ee). Due to the importance of a base for the anionic chiral phase-transfer catalysis, both inorganic and organic bases were examined, and proton sponge (PS) was found to be efficient to increase the yield (Table 2, entry 2). When the reaction temperature was decreased from rt to 0 °C, the reaction proceeded smoothly with increased enantioselectivity (56% to 65% ee) but a prolonged reaction time was needed (Table 2, entry 3). However, the addition of 4 Å molecular sieves (MS) led to a complicated reaction mixture with a decreased yield and enantioselective control (Table 2, entry 4).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entrya | Solvent | Time | Temp. (°C) | Yieldb (%) | eec (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
a Reaction conditions: 1b (0.2 mmol), C6 (0.02 mmol), Selectfluor (0.22 mmol), PS (0.22 mmol) in solvent (4 mL).
b Isolated yield.
c Determined by HPLC analysis.
d Na2CO3 instead of PS.
e 4 Å MS was used.
f C6H5F/CH3CN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2 mL).
g 5 mol% C6. PS: proton sponge. :1, 2 mL).
g 5 mol% C6. PS: proton sponge.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1d | C6H5F | 8 h | rt | 48 | 55 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | C6H5F | 8 h | rt | 72 | 56 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | C6H5F | 16 h | 0 | 60 | 65 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4e | C6H5F | 11 h | 0 | 41 | 5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | CH3CN | 10 min | 0 | 78 | 47 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | C6H5F/CH3CN | 10 min | 0 | 50 | 69 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | C6H5F/CH3CN | 16 h | −60 | 64 | 90 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8f | C6H5F/CH3CN | 16 h | −60 | 87 | 90 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9f,g | C6H5F/CH3CN | 18 h | −60 | 66 | 90 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10d | C6H5F/CH3CN | 18 h | −60 | 71 | 60 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
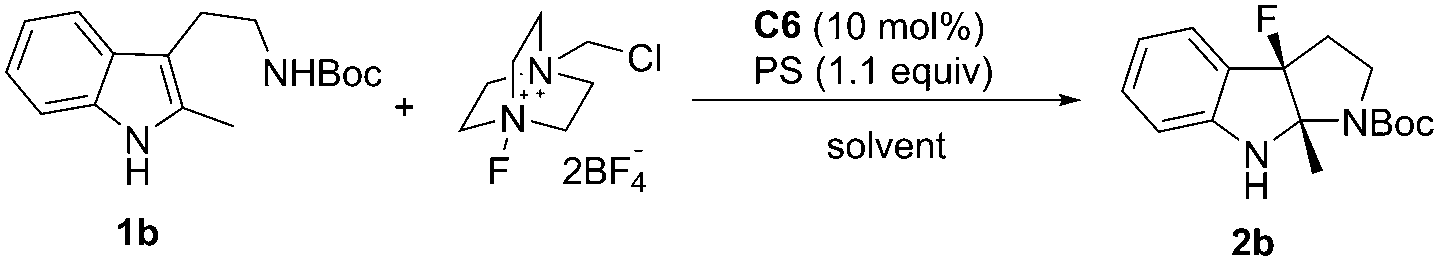
Different from the phenomena where substrates treated with Selectfluor under homogeneous conditions were converted to a complex mixture in the chiral anion phase-transfer catalytic system,4,5 screening polar solvents as homogeneous conditions for this reaction afforded positive results (see the ESI† for details). Among those tested, acetonitrile was the most optimal to provide 2b in 78% yield; however, the enantioselectivity was decreased slightly (Table 2, entry 5). These results encouraged us to test mixed solvents. When mixed solvents of fluorobenzene and acetonitrile (1:1) were utilized, the desired product 2b was obtained in 50% yield and 69% ee within 10 min (Table 2, entry 6). Further decreasing the reaction temperature to −60 °C afforded 2b in 64% yield with 90% ee (Table 2, entry 7). To our delight, running the reaction in a higher concentration improved the yield to a satisfactory level (87% yield) (Table 2, entry 8). Notably, 5 mol% catalyst loading gave the same enantioselectivity (90% ee) and 66% yield (Table 2, entry 9).
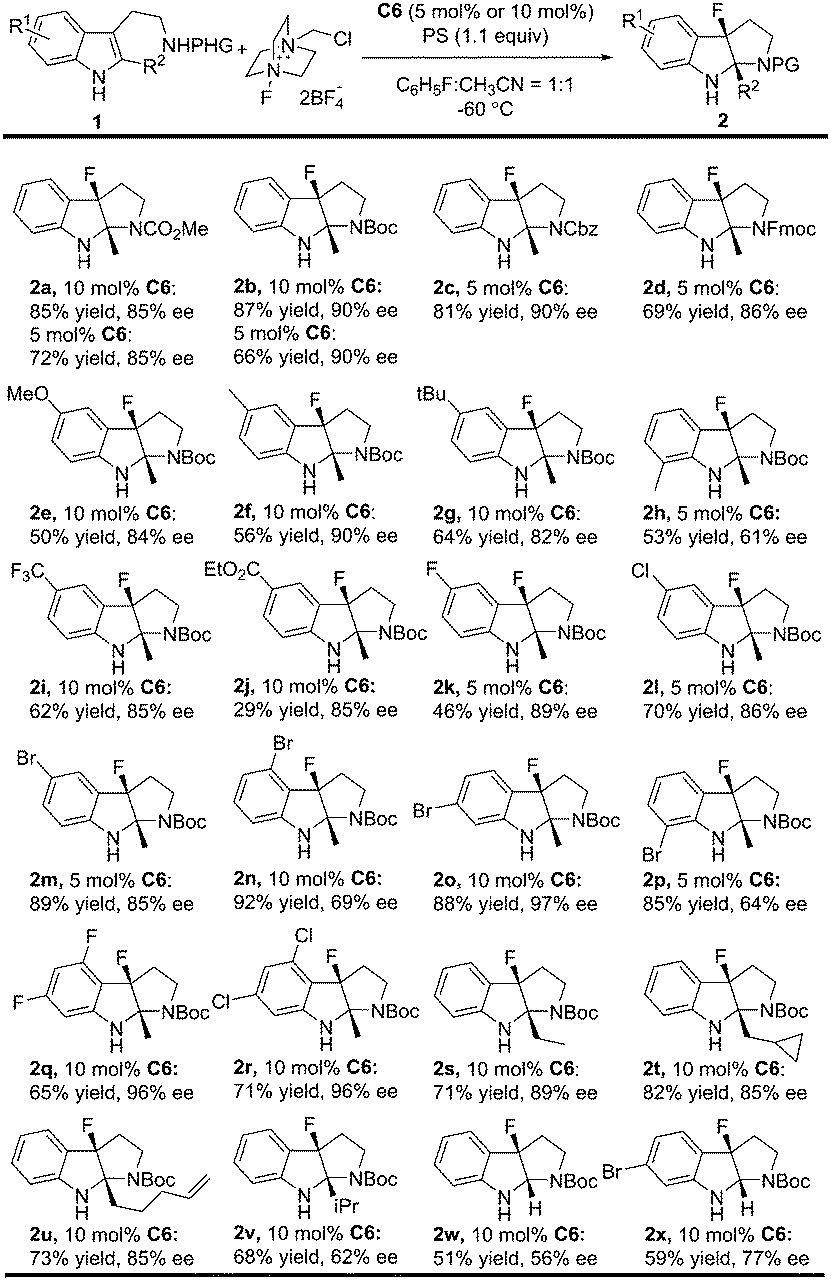
With the optimal reaction conditions in hand (Table 2, entry 8), the substrate scope was explored to test the generality of this asymmetric fluorinative dearomatization reaction. Firstly, the protecting group of the tryptamine was evaluated. As shown in Table 3, in general, all the substrates with electron-withdrawing protecting groups (CO2Me, Boc, Fmoc and Cbz) were converted to their corresponding fluorinative dearomatization products with satisfactory enantioselectivity (85–90% ee, 2a to 2d) and moderate to good yields. The substituents on the indole moiety were further explored. N-Boc protected tryptamines with varied electron-donating substituents (5-CH3, 5-MeO, 5-tBu) at the C5 position were well tolerated to provide their corresponding products with good enantioselectivity (82–90% ee) and 50–64% yields (2e to 2g), and a methyl group at the C7 position led to only moderate enantioselectivity (61% ee) due to the steric hindrance effect (2h). The electron-withdrawing substituent (5-F, 5-Cl, 5-Br, 5-CF3 and 5-CO2Et) at the C5 position of the indole moiety was also well tolerated to provide the target compounds with good enantioselectivity (85–89% ee) and 29–89% yields (2i to 2m). Substrates bearing a Br substituent at the different positions (4, 5, 6, 7) of the indole core underwent dearomatization smoothly (84–92% yields) but with varied enantioselectivity (64–97% ee). Notably, a 6-Br product was obtained in 97% ee (2o). Interestingly, the reactions of 4,6-dihalo-substituted substrates proceeded with excellent enantioselectivity (96% ee, 2q, 2r). Finally, the 2-substituent of the indole moiety was evaluated, and a good enantioselectivity (85–89% ee) was obtained for substrates bearing simple alkyl (2s), cyclopropyl (2t), and alkenyl functional groups (2u). The reaction conditions were also compatible with substrates bearing more hindered substituents or simple H with slightly dropped enantioselectivity (2v, 2w, 2x, 51–68% yields, 56–77% ee).
|
a Reaction conditions: 1 (0.2 mmol), C6 (0.02 mmol), Selectfluor (0.22 mmol), base (0.22 mmol) in C6H5F:CH3CN (1:1, 2 mL) at −60 °C. Isolated yield. ee was determined by HPLC analysis.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Interestingly, the reaction of 3,5-dimethyl substituted substrate 1y under the optimal reaction conditions led to the isolation of 2yy in 72% yield and 93% ee. It is likely that the dearomatized intermediate 2y is unstable due to the introduction of three methyl groups on the indole ring, which facilitates the C–F bond cleavage to result in the formation of a stable benzyl carbenium (Scheme 2).
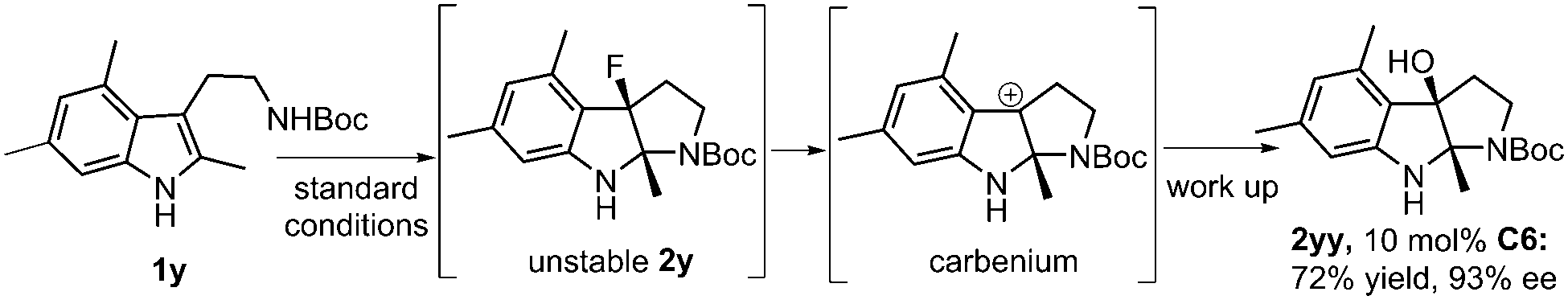 | ||
| Scheme 2 The fluorinative dearomatization reaction of 1y and hydrolysis. | ||
The reaction of 1b in the gram-scale proceeded well affording the dearomative product 2b in 79% yield and 86% ee (Scheme 3). Furthermore, the protection reaction of N–H in 2b with trifluoroacetic anhydride was carried out to give 4 in 88% yield, and the Boc group in 3 could be removed by TMSOTf at −78 °C (Scheme 4). X-Ray crystallography analysis of a single crystal of enantiopure 3 revealed its absolute configuration as 3aR, 8aS. Treatment of 1b with NBS and 1,3,5-trichloroisocyanuric acid (TCCA) led to the dibromo compound 2bb (68% yield, 86% ee) together with 32% yield of the monobromo compound 2p, and the dichloro compound 2bc (66% yield, 86% ee), respectively.
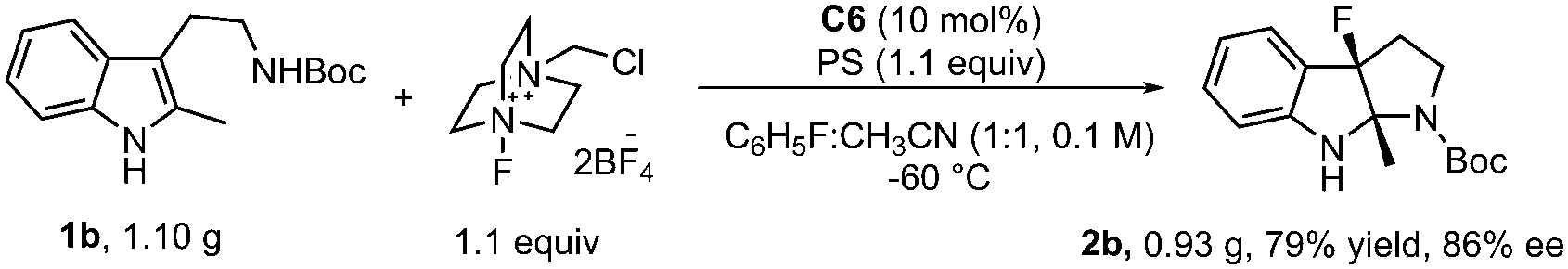 | ||
| Scheme 3 Gram-scale reaction. | ||
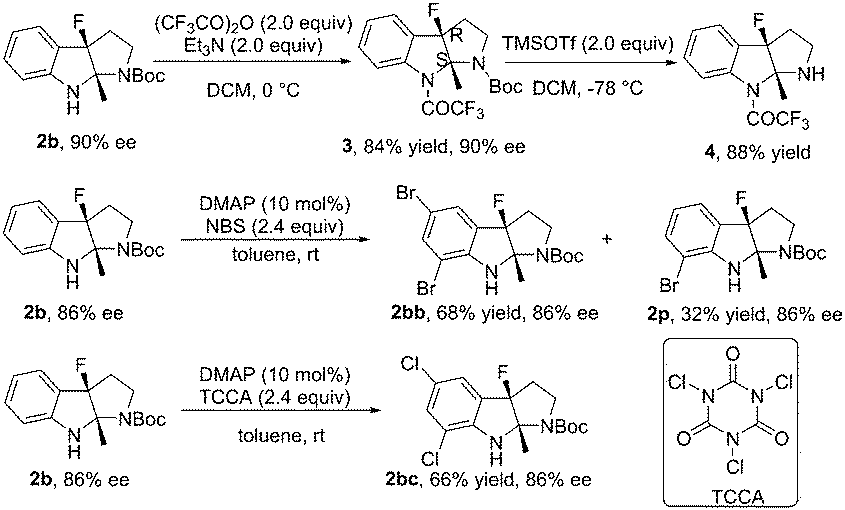 | ||
| Scheme 4 Transformations of product 2b. | ||
To shed light on the reaction mechanism, control experiments were carried out to examine the effects of acid and base additives to the reaction outcomes. The correlation between conversion of 1b with reaction time under varied reaction conditions is shown in Fig. 1. Notably, a strong background reaction was observed. The rate of the reaction of 1b without a chiral phosphoric acid catalyst or PS was similar to that under the standard conditions (entry 8, Table 2). We envisioned that HBF4 released from Selectfluor might accelerate the reaction. Indeed, when 1 equiv. of HBF4 was added, the reaction becomes even faster while the reaction becomes sluggish when 1.2 equiv. of PS was employed. These data imply that the role of PS in this reaction is to neutralize HBF4 generated in situ and hence inhibit the racemic background reaction.
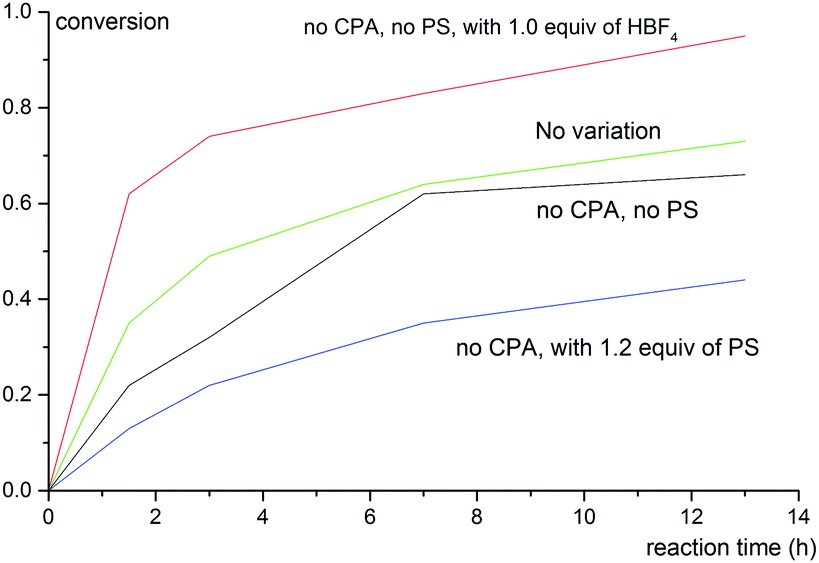 | ||
| Fig. 1 Correlation between conversion of 1b with reaction time under varied reaction conditions. The variation from the “standard reaction conditions” (entry 8, Table 2) is denoted for each plot. | ||
In conclusion, an asymmetric fluorinative dearomatization reaction of tryptamine derivatives was developed based on a chiral anion phase transfer strategy. The preliminary investigations on the reaction mechanism suggested that the reaction proceeds via bifunctional activation by a chiral BINOL-derived phosphate anion. This method is characterized by simple operation, facile introduction of a fluorine atom in a highly enantioselective manner and construction of two contiguous quaternary stereogenic centers.
We thank the National Key Research and Development Program of China (2016YFA0202900), the National Basic Research Program of China (2015CB856600), NSFC (21332009, 21361140373, and 21421091), and the Chinese Academy of Sciences (XDB20000000, QYZDY-SSW-SLH012) for generous financial support.
Notes and references
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, Oxford, UK, 2009 Search PubMed; (c) V. Gouverneur and K. Müller, Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications, Imperial College Press, London, 2012 Search PubMed; (d) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, Germany, 2nd edn, 2013 Search PubMed.
- For recent reviews on catalytic asymmetric fluorination reactions, see: (a) M. C. Pacheco, S. Purser and V. Gouverneur, Chem. Rev., 2008, 108, 1943 CrossRef CAS PubMed; (b) J. R. Wolstenhulme and V. Gouverneur, Acc. Chem. Res., 2014, 47, 3560 CrossRef CAS PubMed; (c) J. Wu, Tetrahedron Lett., 2014, 55, 4289 CrossRef CAS; (d) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (e) T. Sugiishi, M. Matsugi, H. Hamamotob and H. Amii, RSC Adv., 2015, 5, 17269 RSC; (f) P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073 CrossRef CAS PubMed.
- For recent reviews on dearomatization reactions, see: (a) A. R. Pape, K. P. Kaliappan and E. P. Kündig, Chem. Rev., 2000, 100, 2917 CrossRef CAS PubMed; (b) A. Pelter and R. S. Ward, Tetrahedron, 2001, 57, 273 CrossRef CAS; (c) E. P. Kündig and A. Pape, Top. Organomet. Chem., 2004, 7, 71 CrossRef; (d) W. D. Harman, Top. Organomet. Chem., 2004, 7, 95 CrossRef CAS; (e) S. Quideau, L. Pouységu and D. Deffieux, Curr. Org. Chem., 2004, 8, 113 CrossRef CAS; (f) F. López Ortiz, M. J. Iglesias, I. Fernández, C. M. Andújar Sánchez and G. R. Gómez, Chem. Rev., 2007, 107, 1580 CrossRef PubMed; (g) S. Quideau, L. Pouységu and D. Deffieux, Synlett, 2008, 467 CrossRef CAS; (h) L. Pouységu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235 CrossRef; (i) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed; (j) C.-X. Zhuo, C. Zheng and S.-L. You, Acc. Chem. Res., 2014, 47, 2558 CrossRef CAS PubMed; (k) Q. Ding, X. Zhou and R. Fan, Org. Biomol. Chem., 2014, 12, 4807 RSC; (l) W.-T. Wu, L. Zhang and S.-L. You, Chem. Soc. Rev., 2016, 45, 1570 RSC; (m) W. Sun, G. Li, L. Hong and R. Wang, Org. Biomol. Chem., 2016, 14, 2164 RSC; (n) X.-W. Liang, C. Zheng and S.-L. You, Chem. – Eur. J., 2016, 22, 11918 CrossRef CAS PubMed; (o) C. Zheng and S.-L. You, Chem, 2016, 1, 857 CrossRef.
- (a) O. Lozano, G. Blessley, T. M. del Campo, A. L. Thompson, G. T. Giuffredi, M. Bettati, M. Walker, R. Borman and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 8105 CrossRef CAS PubMed . For other examples of fluorinative dearomatization of tryptamine derivatives, see: ; (b) N. Shibata, T. Tarui, Y. Doi and K. L. Kirk, Angew. Chem., Int. Ed., 2001, 40, 4461 CrossRef CAS; (c) T. Fujiwara, T. Seki, T. Yakura and Y. Takeuchi, J. Fluorine Chem., 2014, 165, 7 CrossRef CAS.
- (a) V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681 CrossRef CAS PubMed . For reviews on anionic chiral PTC strategy, see:; (b) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS PubMed; (c) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef CAS PubMed; (d) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534 CrossRef CAS PubMed.
- (a) R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 1268 CrossRef CAS PubMed . For leading examples using the anionic chiral PTC strategy, see: ; (b) R. J. Phipps, K. Hiramatsu and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 8376 CrossRef CAS PubMed; (c) Y.-M. Wang, J. Wu, C. Hoong, V. Rauniyar and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 12928 CrossRef CAS PubMed; (d) T. Honjo, R. J. Phipps, V. Rauniyar and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 9684 CrossRef CAS PubMed; (e) H. P. Shunatona, N. Früh, Y.-M. Wang, V. Rauniyar and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 7724 CrossRef CAS PubMed; (f) A. J. Neel, J. P. Hehn, P. F. Tripet and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 14044 CrossRef CAS PubMed; (g) J. Wu, Y.-M. Wang, A. Drljevic, V. Rauniyar, R. J. Phipps and F. D. Toste, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13729 CrossRef CAS PubMed; (h) A. D. Lackner, A. V. Samant and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 14090 CrossRef CAS PubMed.
- For asymmetric brominative dearomatization reactions of tryptamines, see: (a) W. Xie, G. Jiang, H. Liu, J. Hu, X. Pan, H. Zhang, X. Wan, Y. Lai and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 12924 CrossRef CAS PubMed; (b) X. Feng, G. Jiang, Z. Xia, J. Hu, X. Wan, J.-M. Gao, Y. Lai and W. Xie, Org. Lett., 2015, 17, 4428 CrossRef CAS PubMed . Notably, the asymmetric fluorination of tryptamine derivative 1a under the same catalytic conditions only gave moderate yield and almost racemic result (45% yield, 5% ee).
- For selected reviews on chiral phosphoric acid catalysis, see: (a) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS PubMed; (b) M. Terada, Synthesis, 2010, 1929 CrossRef CAS; (c) J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156 CrossRef CAS PubMed; (d) M. Rueping, A. Kuenkel and I. Atodiresei, Chem. Soc. Rev., 2011, 40, 4539 RSC; (e) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed , For pioneering contributions, see: ; (f) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356 CrossRef CAS PubMed; (g) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566 CrossRef CAS PubMed.
- For selected examples of CADA reactions of indoles, see: (a) Q.-F. Wu, H. He, W.-B. Liu and S.-L. You, J. Am. Chem. Soc., 2010, 132, 11418 CrossRef CAS PubMed; (b) Q.-F. Wu, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 1680 CrossRef CAS PubMed; (c) Q. Yin and S.-L. You, Org. Lett., 2013, 15, 4266 CrossRef CAS PubMed; (d) Q. Yin and S.-L. You, Org. Lett., 2014, 16, 2426 CrossRef CAS PubMed; (e) Q. Cai, Q. Yin and S.-L. You, Asian J. Org. Chem., 2014, 3, 408 CrossRef CAS; (f) D. Duan, Q. Yin, S. Wang, Q. Gu and S. You, Acta Chim. Sin., 2014, 72, 1001 CrossRef CAS; (g) W. Shao, H. Li, C. Liu, D.-J. Liu and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 7684 CrossRef CAS PubMed; (h) C.-X. Zhuo, Y. Zhou, Q. Cheng, L. Huang and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 14146 CrossRef CAS PubMed; (i) X. Zhang, W.-B. Liu, H.-F. Tu and S.-L. You, Chem. Sci., 2015, 6, 4525 RSC; (j) L. Han, W. Zhang, X.-X. Shi and S.-L. You, Adv. Synth. Catal., 2015, 357, 3064 CrossRef CAS; (k) C. Liu, J.-C. Yi, Z.-B. Zheng, Y. Tang, L.-X. Dai and S.-L. You, Angew. Chem., Int. Ed., 2016, 55, 751 CrossRef CAS PubMed; (l) R.-D. Gao, Q.-L. Xu, B. Zhang, Y. Gu and S.-L. You, Chem. – Eur. J., 2016, 22, 11601 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1526408. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc02419c |
| This journal is © The Royal Society of Chemistry 2017 |