
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of single-site copper catalysts for methane partial oxidation†
S.
Grundner
,
W.
Luo
,
M.
Sanchez-Sanchez
* and
J. A.
Lercher
*
Department of Chemistry and Catalysis Research Center, Technische Universität München, Lichtenbergstr. 4, D-85747 Garching, Germany. E-mail: m.sanchez@tum.de; Johannes.lercher@ch.tum.de; Tel: +49 89 289 13540
First published on 24th December 2015
Abstract
Cu-Exchanged zeolites are known as active materials for methane oxidation to methanol. However, understanding of the formation of Cu active species during synthesis, dehydration and activation is fragmented and rudimentary. We show here how a synthesis protocol guided by insight in the ion exchange elementary steps leads to highly uniform Cu species in mordenite (MOR).
Cu-Exchanged zeolites have attracted broad attention since the discovery of their high NO decomposition activity in 1986,1 and their high selectivity for the partial oxidation of methane to methanol in 2005.2 Numerous publications on the speciation of Cu in zeolites with different topologies exemplify the relevance of the structure of the Cu ions and/or Cu–O clusters for catalytic applications.3–5 However, the formation of Cu spectators, often quantitatively more abundant than the active species, hampers the identification of active sites and impedes a rigorous comparison of activities among Cu–zeolite materials from different sources.
Commonly, the Na-exchanged form of zeolites is used for the preparation of transition metal exchanged zeolite catalysts, because of the more straightforward control of the synthesis parameters. However, we have recently obtained significantly higher methane oxidation activities than previous reports, when Cu was exchanged on H-MOR instead of on Na-MOR.6,7 It should be noted in passing that also other authors have reported remarkable differences in the activity of Cu-MOR in presence of Na+ in the zeolite structure.8
In this contribution, we explore, therefore, the impact of gradual Cu2+ ion exchange on the formation of charged Cu oxide (Cu–oxo) complexes during thermal activation in Cu-exchanged mordenite. The effect of alkali and alkaline earth cations (co-cations) and the pH during ion exchange were also quantitatively explored in order to control the formation and nuclearity of Cu–oxo clusters active for methane partial oxidation at low temperatures.
Cu-MOR with different Cu/Al ratios were prepared by ion exchange of H-MOR or Na-MOR respectively with Cu2+ in aqueous phase at ambient temperature. The Brønsted acidic form of MOR, H-MOR, was obtained before by calcination of zeolite NH4-MOR (Clariant, Si/Al = 11) in synthetic air, while Na-MOR was synthesized by exchange of H-MOR with NaNO3. Independently of the anion, identical Cu2+ exchange degrees and methane oxidation activities were obtained as long as the solution was buffered to a pH of 5.7 during ion exchange. The zeolite was also exchanged with Cu2+ ions in the presence of other cations (Na+, K+, Mg2+, Ca2+). The characterization of these materials is compiled in the ESI† (see Fig. S1 and Table S1). The concentration of active sites in Cu zeolites was quantified by the amount of products collected after a three-stage catalytic oxidation of methane as described in ESI† and in ref. 6. In this way, the specificity of ion exchange protocols for the sites responsible for oxidation of methane was evaluated (Fig. 1 and 2). In Fig. 1, it is seen that Cu2+ ion exchange of H-MOR (CuH-MOR) yields the material with the highest activity per Cu in the in the zeolite. The stoichiometry of three Cu atoms to one methane molecule converted allowed us to conclude that a trinuclear Cu–oxo cluster was formed as active site in MOR.6 The presence of alkali cations (Na, K) or alkaline earth cations (Mg, Ca) drastically reduced this Cu efficiency to values of 5 Cu per oxidized methane, without affecting the selectivity to methanol (in the range 75–80% for all samples shown in Fig. 1).
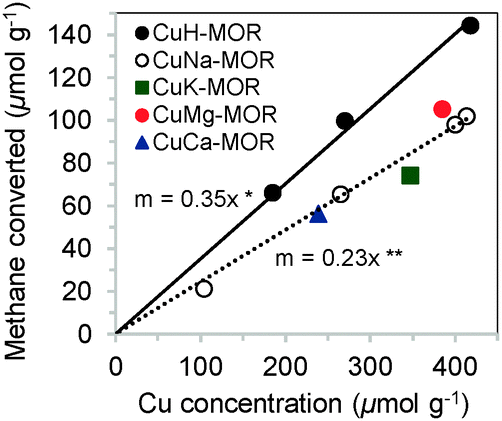 | ||
Fig. 1 Total methane yield versus Cu2+ concentration for Cu-exchanged MOR with various co-cations. *![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The slope of 0.35 indicates that ca. 3 Cu atoms are involved in the oxidation of one methane molecule. The slope of 0.35 indicates that ca. 3 Cu atoms are involved in the oxidation of one methane molecule. | ||
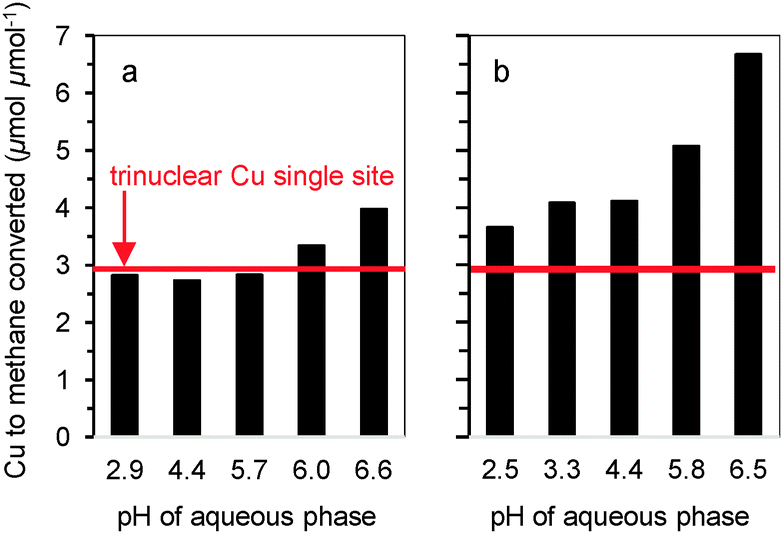 | ||
| Fig. 2 Impact of pH during Cu-exchange on methane oxidation activity of MOR. Cu to oxidized methane ratio of (a) CuH-MOR and (b) CuNa-MOR. | ||
While it has been experimentally established that the Cu2+ site active for partial methane oxidation is a multinuclear Cu site, oxide clusters containing more than three Cu cations show low selectivity towards methanol.6,7,9,10 Thus, as a first boundary condition to form sites for methane partial oxidation selectively, the pH of ion exchange must be kept below 7 to avoid precipitation of Cu(OH)2.11 Furthermore, the point of zero charge of the zeolite surface silanol groups is 6.0. Near and above this pH the deprotonation of SiOH and consequently the exchange with Cu2+ will occur,12 which also leads to the formation of nano-sized CuO particles during calcination. The formation of such undesirable CuO species was detected by transmission electron microscopy (see Fig. S2 in the ESI†) for those samples prepared exceeding the upper pH limit of 6.0. On the other hand, to maximize the formation of copper active dimers and/or trimers, a high concentration of Brønsted acid sites (BAS) must be exchanged with Cu2+. Therefore, also too low pH is not ideal, because it reduces the exchangeability of BAS.
Considering these boundary conditions, ion exchange of BAS protons was maximized without formation of large copper oxide species by buffering the exchange solution to pH 5.7. Hydrolysis and back-exchange (with NaNO3) was used to probe quantitatively the nature of the Cu species. All Cu (irrespective of whether it is a monomer, a dimer or a trimer) coordinated to framework Al tetrahedra is readily exchangeable with other mono- and divalent cations in water. In contrast, CuO or Cu(OH)2 nanoparticles or clusters cannot be hydrolyzed and are not back-exchanged in consequence. This allows the quantitative differentiation of CuO or Cu(OH)2 from the ion exchanged species by chemical analysis of the material after back-exchange. Since all Cu2+ was back-exchanged for Na+ in Cu-MOR samples prepared at pH < 6 (see Table S2 and Section S2 in the ESI†), we are able to rule out the presence of surface copper oxide aggregates. Thus, the efficiencies reported in Fig. 1 are solely related to well-defined Cu species in the zeolite micropores.
The constant ratio of methane oxidized in relation to Cu present in CuH-MOR indicates that this synthetic approach leads to the formation of a single site catalyst. The stoichiometry of Cu to activated methane of 3:1 in all CuH-MOR materials is in accordance with the previously established single-site trinuclear copper oxygen cationic cluster in MOR for oxidation of methane to methanol.6 The presence of other cations did not affect the selectivity to methanol but caused a lower concentration of Cu in such sites (lower Cu efficiency), i.e., about 25% of Cu sites became inactive (Fig. 1). In order to understand the role of other cations hindering the formation of the Cu active species, a series of CuH-MOR and CuNa-MOR materials were prepared by ion exchange at different pH, but with the same Cu concentrations (0.01 M). There were no significant changes in the selectivity to form these cationic clusters for Cu-exchanged H-MOR prepared in the pH range 2–6 (Fig. 2a), i.e., all Cu was present in trinuclear Cu–oxide clusters. In contrast, the pH during Cu2+ exchange on Na-MOR had a significant impact on the activity for methane oxidation (Fig. 2b). Cu efficiency decreased substantially with increasing pH, pointing to Cu cations not participating in methane oxidation. The loading of Cu (active and inactive) increased with increasing pH regardless of the co-cation. It is hypothesized that this increase is associated with the shift of the hydrolysis equilibrium from divalent [Cu(H2O)6]2+ to monovalent [Cu(H2O)5OH]+.13 This indicates that the latter species is predominantly exchanged in aqueous ion-exchange of zeolites at relatively high pH values.14
The high uniformity of Cu species formed in CuH-MOR in a broad range of pH is in contrast to the heterogeneity of Cu species that is inferred from the different Cu to methane stoichiometries, observed when co-cations are present. The self-organization of Cu2+ to a uniform oxygen containing cluster in H-MOR points to a high mobility of Cu2+ during thermally induced dehydration and oxidation. The hydrated Cu2+ cations predominant at low pH ([Cu(H2O)6]2+), which balance the negative charge of two Al–oxygen tetrahedra, are transformed by dissociation of one water molecule to [Cu(H2O)xOH]+, which balances the negative charge of only one Al–oxygen tetrahedron.15 The simultaneously formed H+ also balances the negative charge of one Al–oxygen tetrahedron. Infrared spectra of adsorbed pyridine as well as adsorption and desorption of NH3 confirmed this hypothesis by quantitatively showing the formation of one BAS per incorporated Cu2+ for a series of CuNa-MOR exchanged at pH 4 (see Fig. S3 and S4 in the ESI†). Consequently, regardless of the hydrolysis equilibrium in aqueous phase, a population of monovalent [Cu–OH]+ species attached to framework Al sites are formed during thermal activation. Such species have the potential to transform to multinuclear Cu–oxo clusters via condensation with an adjacent [Cu–OH]+ forming a μ-oxo bridge between Cu2+ cations.16,17
In order to understand the lower concentrations of Cu sites active for the selective oxidation of methane in CuNa-MOR compared to CuH-MOR and their consistently lower concentration as the pH increases, the location of Na+ in the parent Na-MOR and its exchange for Cu2+ was investigated in detail. The initial position of Na+ and the position of Cu2+ after ion exchange are given by the location of the exchange sites. The concentration of these sites was determined by chemical analysis of Na+ content on a fully exchanged Na-MOR. This value is correlated to the total intensity of the OH band characteristic of BAS in a Na+ free H-MOR. For MOR, this IR band is composed of two contributions, because the wave-number of the O–H stretching band is slightly different for protons in the 12-MR main channels and for protons in the 8-MR pockets. Finally, the concentration of protons in each of these locations was quantified via adsorption of hexane and pyridine, which are probe molecules with different accessibility to the sites (see Table S3 in the ESI†).6,18
The coordination sites occupied by Cu2+ and Na+ were deduced by determining the concentration and location of the bridging OH groups before (Fig. 3a) and after the ion exchange with Cu2+ and Na+ leading to Cu-, Na- and CuNa-MOR. After exchange of H-MOR with Cu2+ and subsequent activation in O2, only H+ located in the 8-MR pore mouth of the side pockets in MOR were consumed (Fig. 3b and Fig. S5 in the ESI†). In addition, Na+ was found to exchange preferentially at sites in the side pockets (Fig. 3c), in good agreement with previous reports.19 Thus, it is concluded that independent of the location of the cations during ion exchange, in the final stage of dehydration and thermal activation Na+ is competing with Cu2+ for framework Al–O tetrahedra located in the side pockets. Therefore, we attribute the lower activity of CuNa-MOR compared to CuH-MOR to a competition of Na for those T-sites required to host the active Cu–oxo cluster.
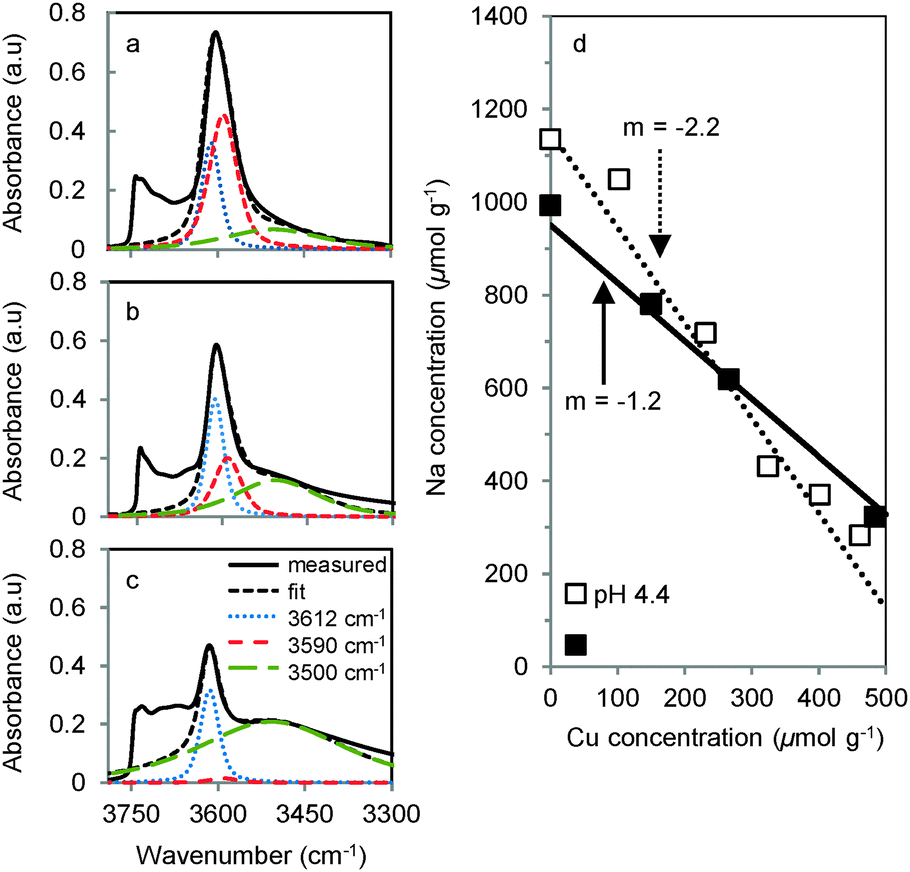 | ||
| Fig. 3 Deconvoluted infrared spectra of the OH stretching vibrations of (a) H-MOR, (b) Cu-HMOR with Cu/Al 0.3 and (c) Na-MOR with Na/Al 0.6 after activation at 450 °C. BAS are deconvoluted in bands attributed to main channel OH (3612 cm−1), side pocket OH (3590 cm−1) and perturbed OH stretching vibrations (3500 cm−1). (d) Na+ concentrations in CuNa-MOR prepared at different pH as a function of Cu loading. | ||
When exchanging Cu2+ on Na-MOR the Na+ concentration in the zeolite decreased linearly with increasing Cu2+ loading, which demonstrates that all Cu2+ introduced into the samples gradually exchanged for Na+ at the preferred 8-MR sites (Fig. 3d). The slope of 2.2 for the series prepared by ion exchange at pH 4.4, indicates that one Cu2+ substituted two Na+ cations. This confirms the formation of [Cu(H2O)6]2+ species balancing the negative charges of two Al–O tetrahedra. In contrast, Cu-exchanged Na-MOR prepared at pH 5.7 showed a slope of 1.2 indicating that a monovalent [Cu(H2O)5OH]+ species is replacing one Na+ at one framework Al–O tetrahedron. Therefore, the relative higher catalytic efficiency of CuNa-MOR samples prepared at low pH is attributed to the exchange of two Na+ for divalent [Cu(H2O)6]2+ allowing a more efficient elimination of competing Na+ cations.
Active Cu clusters currently proposed in literature consist of 2 or 3 Cu atoms interconnected by μ-oxo bridges and attached to two framework Al in the 8-MR pore mouth of the side pockets.6,20 It has been observed by IR that the protons present, e.g., in CuNa-MOR samples are mainly located in the 12-MR channel (see Table S4 in the ESI†). Thus, the concentrations of co-cations left occupy the exchange positions in the side pockets that are not occupied by Cu species. This is illustrated in Fig. 4, where the methane converted is represented vs. the concentration of Cu2+ and of Na+ in the side pockets (details are given in the ESI†). The competition of Na+ (or other co-cations) and Cu2+ for the thermodynamically most stable cationic positions in MOR reduces the statistical occupancy of vicinal Cu2+ in the side pockets and, therefore, the probability of [CuOH]+ exchanged species to form a multinuclear Cu–oxo-site. While in Fig. 1 it could be seen that samples containing the same concentration of Cu (ca. 400 μmol g−1) were significantly less active if a co-cation is present, in Fig. 4 it is clear that such effect is related to the presence of different concentrations of Na+ in the side pockets. In other words, the decrease of the activity observed in CuX-MOR samples (X = Na, K, Mg and Ca) is caused by the formation of inactive isolated Cu2+ in those side pockets that are partially occupied by Na+. Thus, we hypothesize that the formation of active Cu–oxo clusters requires the exchange of at least two Cu ions in two Al sites of the same 8-MR. For the formation of the highly efficient [Cu3O3]2+ cluster reported for CuH-MOR, a third Cu in the vicinity is required to react with the two Cu2+ located in the side pockets. We conclude, therefore, that the presence of other cations significantly decreases the probability of self-assembly of di- and trinuclear copper oxo clusters active for methane oxidation. It has been observed that the active Cu–oxo clusters are redistributed to an inactive state during methanol extraction. Thus, even controlling the synthesis of single active sites in Cu-MOR, would not suffice, if the sites do not reform during regeneration. Activity in partial oxidation of methane was measured, therefore, for several consecutive cycles with CuNa-MOR and CuH-MOR materials (see Fig. S6 in the ESI†). Despite the hydrolysis of the active site and the expected mobility of Cu2+ exchanged species upon contact with water, the catalytic activity remained stable over 3 cycles. Thus, an identical concentration of active sites is formed in each subsequent thermal activation. This indicates that the final sitting of Na+ and Cu2+ in dehydrated MOR is thermodynamically determined and only depends on the concentration of both cations in the exchanged material.
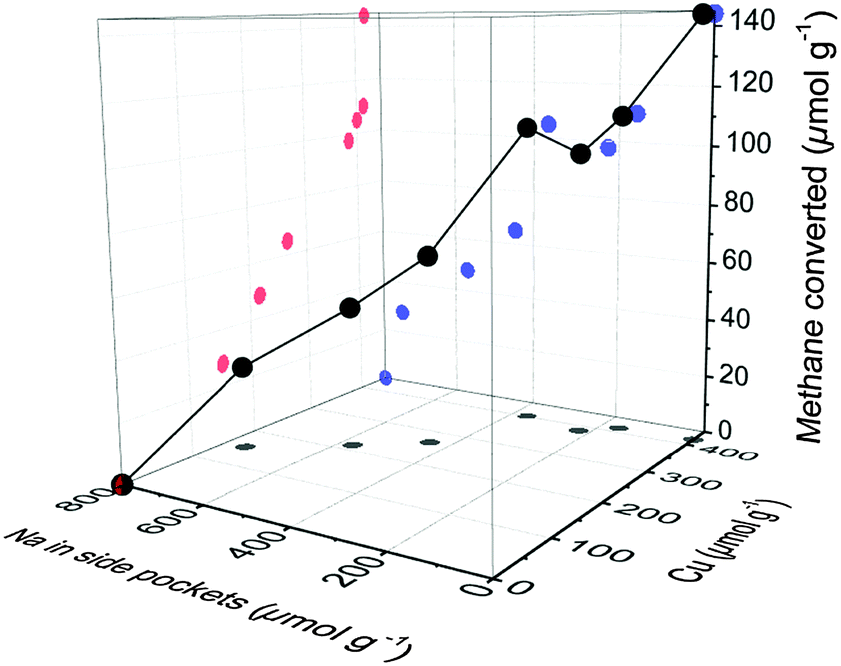 | ||
| Fig. 4 Methane converted as a function of Cu concentration and Na concentration in the 8-MR side pockets. | ||
From the results presented here, the remarkably higher selectivity to form cationic trinuclear Cu–oxo clusters and the constant methanol yield for samples prepared from H-MOR is attributed to the absence of elements hindering the formation of the active clusters. Formation of unselective CuO large clusters was avoided by limiting the pH below the point of zero charge of the zeolite surface silanol groups. The presence of Na+ or other alkali and alkaline earth cations competing for the exchange positions preferred by Cu2+ leads to a heterogeneous speciation of Cu. Depending on the abundance of the co-cations in the side pockets, tri-, di or mononuclear Cu–oxo sites will be formed. Cu monomers are inactive in methane oxidation, while both μ-oxo Cu2O and Cu3O3 clusters have been identified as active sites.6,9 In fact, the presence of co-cations competing for the 8-MR side pocket sites would statistically shift the speciation of Cu towards dimers and inactive monomers. This model explains on the one hand the lower Cu efficiency of catalysts typically prepared with Na-MOR as starting material and, on the other hand, that in situ spectroscopy studies on such catalysts detected only a majority of Cu2O sites.2,10,20
The research was partly supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences under Award DE-SC0012702. It was also supported by the EU NEXT-GTL (Innovative Catalytic Technologies & Materials for Next Gas to Liquid Processes) project.
Notes and references
- M. Iwamoto, H. Furukawa, Y. Mine, F. Uemura, S. I. Mikuriya and S. Kagawa, J. Chem. Soc., Chem. Commun., 1986, 16, 1272 RSC.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394 CrossRef CAS PubMed.
- J. H. Kwak, T. Varga, C. H. F. Peden, F. Gao, J. C. Hanson and J. Szanyi, J. Catal., 2014, 314, 83 CrossRef CAS.
- M. H. Groothaert, J. A. van Bokhoven, A. A. Battiston, B. M. Weckhuysen and R. A. Schoonheydt, J. Am. Chem. Soc., 2003, 125, 7629 CrossRef CAS PubMed.
- J. Dedecek, Z. Sobalik, Z. Tvaruzkova, D. Kaucky and B. Wichterlova, J. Phys. Chem., 1995, 99, 16327 CrossRef CAS.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef CAS PubMed.
- P. J. Smeets, M. H. Groothaert and R. A. Schoonheydt, Catal. Today, 2005, 110, 303 CrossRef CAS.
- K. Narsimhan, V. K. Michaelis, G. Mathies, W. R. Gunther, R. G. Griffin and Y. Roman-Leshkov, J. Am. Chem. Soc., 2015, 137, 1825 CrossRef CAS PubMed.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908 CrossRef CAS PubMed.
- E. M. C. Alayon, M. Nachtegaal, E. Kleymenov and J. A. van Bokhoven, Microporous Mesoporous Mater., 2013, 166, 131 CrossRef CAS.
- L. Hidmi and M. Edwards, Environ. Sci. Technol., 1999, 33, 2607 CrossRef CAS.
- M. Schreier, S. Teren, L. Belcher, J. R. Regalbuto and J. T. Miller, Nanotechnology, 2005, 16, 582 CrossRef CAS PubMed.
- M. Lo Jacono, G. Fierro, R. Dragone, X. Feng, J. d'Itr and W. K. Hall, J. Phys. Chem. B, 1997, 101, 1979 CrossRef CAS.
- M. Iwamoto, H. Yahiro, K. Tanda, N. Mizuno, Y. Mine and S. Kagawa, J. Phys. Chem., 1991, 95, 3727 CrossRef CAS.
- A. E. Hirschler, J. Catal., 1963, 2, 428 CrossRef CAS.
- S. C. Larsen, A. Aylor, A. T. Bell and J. A. Reimer, J. Phys. Chem., 1994, 98, 11533 CrossRef CAS.
- J. Valyon and W. K. Hall, J. Phys. Chem., 1993, 97, 7054 CrossRef CAS.
- F. Eder, M. Stockenhuber and J. A. Lercher, J. Phys. Chem. B, 1997, 101, 5414 CrossRef CAS.
- V. A. Veefkind, M. L. Smidt and J. A. Lercher, Appl. Catal., A, 2000, 194, 319 CrossRef.
- P. Vanelderen, B. E. R. Snyder, M.-L. Tsai, R. G. Hadt, J. Vancauwenbergh, O. Coussens, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2015, 137, 6383 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, elemental analysis, BET analysis, TEM images and acid site characterization of Cu-exchanged MOR samples. See DOI: 10.1039/c5cc08371k |
| This journal is © The Royal Society of Chemistry 2016 |