
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A double hetero[4]helicene composed of two phenothiazines: synthesis, structural properties, and cationic states†
Daisuke
Sakamaki
*a,
Daisuke
Kumano
b,
Eiji
Yashima
b and
Shu
Seki
 *a
*a
aDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Kyoto 615-8510, Japan. E-mail: sakamaki.daisuke.68u@st.kyoto-u.ac.jp; seki@moleng.kyoto-u.ac.jp
bDepartment of Molecular Design and Engineering, Graduate School of Engineering, Nagoya University, Nagoya, Aichi 464-8603, Japan
First published on 5th October 2015
Abstract
A novel double hetero[4]helicene consisting of two phenothiazines (1) has been synthesized. The racemization barrier of 1 is high enough for optical separation. We successfully obtained the single crystals of the radical cation salt of 1˙+, whose torsion angles were decreased compared to those of the neutral state.
Phenothiazine derivatives are an important class of π-electron donors because of their low oxidation potentials and high stability of oxidized species, and have been utilized as the redox-active units or spin sources in studies of organic mixed-valence systems and molecular magnetism. To date, various kinds of phenothiazine oligomers connected through different bridging units have been synthesized and the electronic properties depending on the bridging unit have been investigated.1–4 In particular, directly connected phenothiazine oligomers, which are the simplest multi-redox systems consisting of phenothiazines, have been of interest in the studies of electronic and magnetic interactions among the phenothiazine units.5–11 Another important feature of phenothiazines is the controllability of the structure by redox stimuli. Phenothiazine derivatives possess a bent structure along the S–N axis with the bending angle (denoted as θb) of typically 150–160°, and upon oxidation, the phenothiazine skeleton transforms to the more planar structure.2,8 Therefore, phenothiazines are potential components of redox-active molecular machines.12
Recently, we reported a novel facile synthetic method of double heterohelicenes13 by using the tandem oxidation reaction of N-substituted heteroacenes.14 In the report, we demonstrated the preparation of two kinds of double hetero[5]helicenes from 6,13-dihydro-6,13-diazapentacene and 13H-dibenzo[b,i]phenoxazine via cruciform heteroacene-dimers. This protocol is considered to be versatile approach to obtain double helicenes from other heteroacenes having a NH group. In this study, we applied this strategy to the construction of a novel double hetero[4]helicene by using phenothiazines as building blocks.
At first, phenothiazine-dimers were synthesized with a single C–N connection by the already reported dimerization reaction of phenothiazines using a mixture of dimethylsulfoxide (DMSO) and acetic anhydride as the oxidant.5 In this work, we used 3,7-di-tert-butylphenothiazine (3) as the starting material to prevent the reaction on the most reactive 3 and 7 positions, and obtained the desired 1,10′-dimer in 65% yield. However, this reaction requires a long reaction time (about a week). Next, the dimerization of 3 was examined by DDQ oxidation according to our previous work.14 This approach further improved the yield of 2 up to 78%, as well as provided a short reaction time of several hours (Scheme 1). The oxidation of 2 by a combination of DDQ and Sc(OTf)3 resulted in the intramolecular C–N bond formation and gave a doubly fused phenothiazine dimer 1. Furthermore, 1 was also obtained in an one-pot manner by using the tandem oxidation of 3 with DDQ followed by DDQ/Sc(OTf)3.
 | ||
| Scheme 1 Synthesis of phenothiazine dimers 1 and 2. | ||
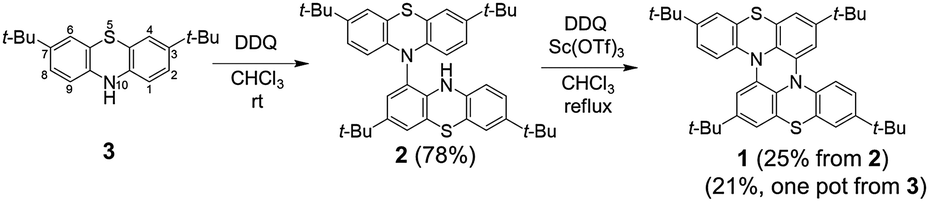
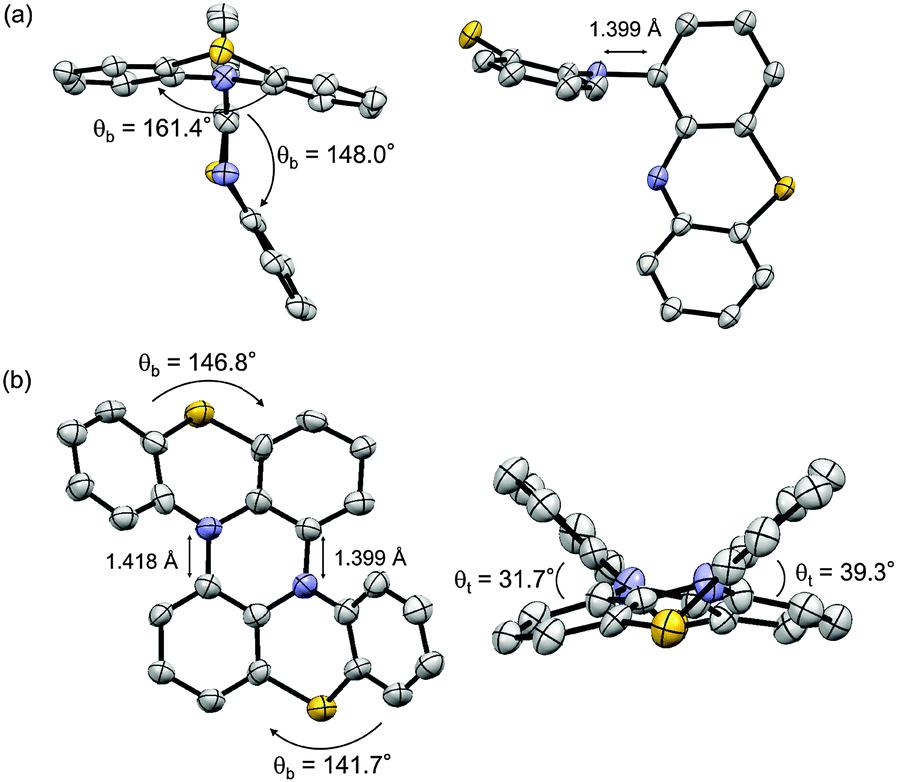
The structures of 2 and 1 were determined by X-ray single crystal analysis (Fig. 1). In the crystal state, 2 took a cruciform structure similar to heteropentacene dimers,14 although both of the phenothiazine units were bent along the S–N axes. As expected, 1 took a double hetero[4]helicene structure, and in a unit cell, two enantiomers ((P,P′)- and (M,M′)-isomers) existed in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The torsion angles in the fjord regions (denoted as θt) were 31.7° and 39.3°, which were larger than those of carbo[5]helicene (ca. 30°),15 reflecting the bent structure of the neutral phenothiazine molecule. The (P,P′)- and (M,M′)-isomers could interconvert via an achiral isomer (mesomer). The energy diagram of this interconversion was calculated using DFT calculations on the model compound 1′ without tBu groups at the B3LYP/6-31G* level (Fig. S8, ESI†).16 The step with the highest barrier is the transformation from the chiral to achiral isomers, and the barrier height was calculated to be 118.0 kJ mol−1. This value is remarkably high compared to the calculated value of dibenzo[a,j]perylene, which corresponds to the all-carbon analogue of 1′ (41.5 kJ mol−1, Fig. S9, ESI†), due to the bent nature of phenothiazine.17,18 In fact, we achieved the optical resolution of 1 by HPLC using a chiral stationary phase. The obtained two fractions showed mirror-image circular dichroism (CD) spectra (Fig. 2). Spectral simulations based on time-dependent DFT (TD-DFT) calculations predicted the faster eluting fraction as the (P,P′)-isomer, and the slower one as the (M,M′)-isomer, respectively (Fig. S20, ESI†). The decay of the CD spectra of 1 obeyed first-order kinetics, and the racemization barrier was determined to be 112.8 kJ mol−1 by the Arrhenius plot of the rate constants at 30, 40, 50, and 60 °C (Fig. S7, ESI†). This value was in good accordance with the theoretical calculations, and higher than that of carbo[5]helicene (101 kJ mol−1).19 We also carried out the theoretical calculations for the analogues of 1 having O and Se instead of S atoms, and found that the analogue with heavier chalcogen atoms has larger torsion angles and a higher inversion barrier (Fig. S10 and S11, ESI†).
:1. The torsion angles in the fjord regions (denoted as θt) were 31.7° and 39.3°, which were larger than those of carbo[5]helicene (ca. 30°),15 reflecting the bent structure of the neutral phenothiazine molecule. The (P,P′)- and (M,M′)-isomers could interconvert via an achiral isomer (mesomer). The energy diagram of this interconversion was calculated using DFT calculations on the model compound 1′ without tBu groups at the B3LYP/6-31G* level (Fig. S8, ESI†).16 The step with the highest barrier is the transformation from the chiral to achiral isomers, and the barrier height was calculated to be 118.0 kJ mol−1. This value is remarkably high compared to the calculated value of dibenzo[a,j]perylene, which corresponds to the all-carbon analogue of 1′ (41.5 kJ mol−1, Fig. S9, ESI†), due to the bent nature of phenothiazine.17,18 In fact, we achieved the optical resolution of 1 by HPLC using a chiral stationary phase. The obtained two fractions showed mirror-image circular dichroism (CD) spectra (Fig. 2). Spectral simulations based on time-dependent DFT (TD-DFT) calculations predicted the faster eluting fraction as the (P,P′)-isomer, and the slower one as the (M,M′)-isomer, respectively (Fig. S20, ESI†). The decay of the CD spectra of 1 obeyed first-order kinetics, and the racemization barrier was determined to be 112.8 kJ mol−1 by the Arrhenius plot of the rate constants at 30, 40, 50, and 60 °C (Fig. S7, ESI†). This value was in good accordance with the theoretical calculations, and higher than that of carbo[5]helicene (101 kJ mol−1).19 We also carried out the theoretical calculations for the analogues of 1 having O and Se instead of S atoms, and found that the analogue with heavier chalcogen atoms has larger torsion angles and a higher inversion barrier (Fig. S10 and S11, ESI†).
 | ||
| Fig. 1 X-ray structure of (a) 2 and (b) 1. Thermal ellipsoids are set at 50% probability. The tBu groups and hydrogen atoms were omitted for clarity. | ||
 | ||
| Fig. 2 CD and absorption spectra of the faster and the more slowly eluting enantiomers of 1 in CH2Cl2 at 298 K. | ||
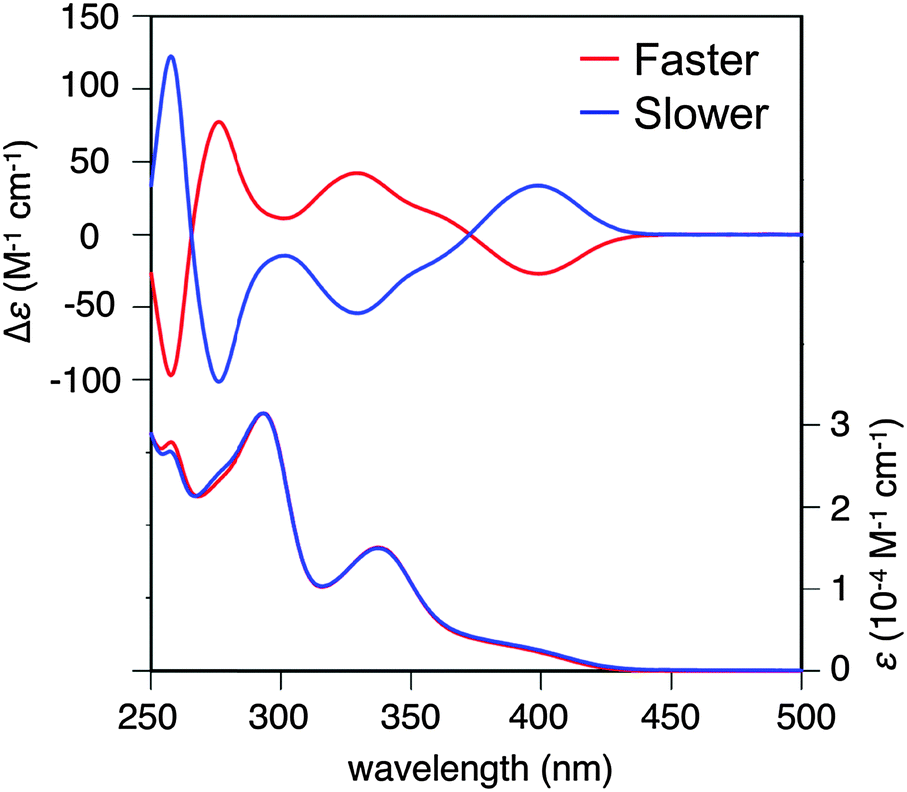
Because 1 and 2 are regarded as dual-electron donor molecules, herein we focus on HOMO and (HO−1)MO. For 2′, HOMO and (HO−1)MO were energetically close with an energy gap of 0.125 eV and completely localized on one side of the two phenothiazine units, resulted from their perpendicular connection (Fig. 3a). On the other hand, for 1′, HOMO and (HO−1)MO were totally delocalized over the whole molecular skeleton, and the energy gap between these two MOs was increased to 0.613 eV (Fig. 3b).
 | ||
| Fig. 3 Frontier Kohn–Sham MOs of (a) 2′ and (b) 1′ at the B3LYP/6-31G* level. | ||
The redox properties of 1 and 2 were investigated via cyclic voltammetry measurements (Fig. S13, ESI†).20 For 2, the consecutive oxidation processes of E1 = 0.187 V and E2 = 0.296 V (vs. ferrocene/ferrocenium) were observed, and the potential difference ΔE = E2 − E1 was only 0.109 V, stemming from little electronic communication between two phenothiazine moieties of 2. For 1, on the other hand, the potentials of E1 (0.023 V) and E2 (0.655 V) markedly separated and ΔE was significantly increased to 0.632 V. This large ΔE value clearly indicates the increased electronic communication between two phenothiazine moieties in 1 by ring-fusion. The E1 value of 1 was lower than that of 2 by 0.164 V, suggesting the good electron donor properties of 1.
The UV-Vis-NIR absorption spectral changes of 1 during electrochemical oxidation were measured (Fig. 4). Upon oxidation, a low energy absorption band with a peak top at 0.83 eV (1500 nm) appeared, in addition to the band at 2.28 eV (545 nm) corresponding to the absorption of the radical cation of phenothiazines.8,21 The low energy band in the NIR region could be ascribed to the charge-resonance band between two fused phenothiazine units, judging from the TD-DFT calculations of 1′˙+ (Fig. S16, ESI†).22 By applying higher voltage, the spectral shape was continuously changed with isosbestic points, according to the simple 1 electron oxidation process from 1˙+ to 12+ without the formation of any significant byproducts.
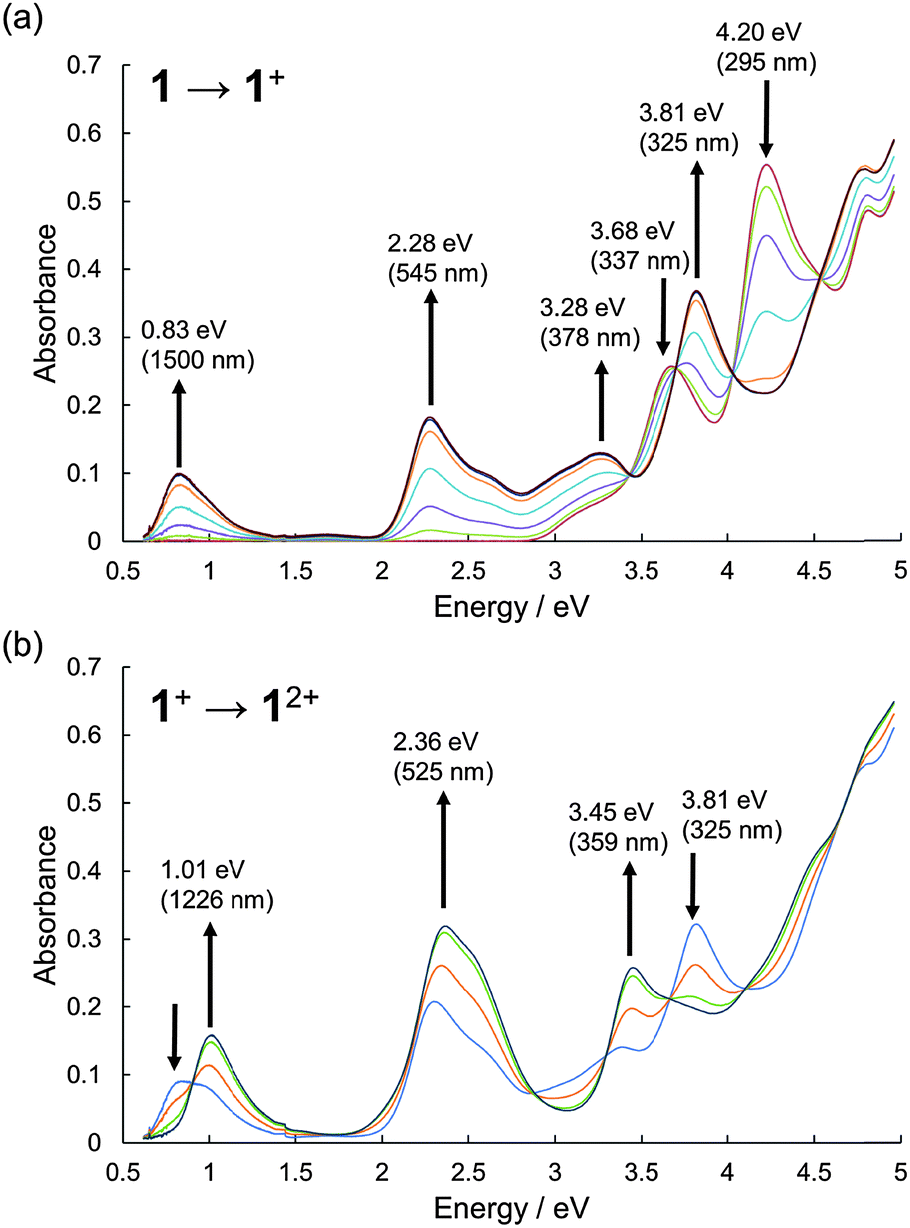 | ||
| Fig. 4 UV-Vis-NIR absorption spectra during the electrochemical oxidation of 1. (a) 1 to 1˙+ and (b) 1˙+ to 12+. The spectra were measured in CH2Cl2 with 0.1 M n-Bu4NBF4 at 298 K. | ||
Motivated by the results of electrochemical measurements, we tried to isolate cation salts of 1 and 2 by chemical oxidation. The single crystals of a radical cation salt of 1˙+ was obtained as red-purple needles by slow evaporation of a CH2Cl2/hexane solution of 1 oxidized with 1 equiv. of tris(4-bromophenyl) aminium hexachloroantimonate (magic blue). The X-ray crystal analysis revealed the structure of 1˙+ (Fig. 5). The dihedral angles in the fjord regions of 1˙+ were smaller compared with those of the neutral 1. This shrinkage of the helical pitches are ascribed to the increase of planarity of two phenothiazine units upon oxidation.17,18 The average length of the two C–N bonds connecting two phenothiazine moieties also became shorter than that of 1. In the crystal of 1˙+SbCl6−, a SbCl6− anion existed on a saddle-shaped 1˙+, as shown in Fig. 5c. The radical cation 1˙+ is stable under ambient conditions both in the solution and the solid state, and the absorption spectrum of the CH2Cl2 solution remained unchanged after 2 weeks under air and room light (Fig. S17, ESI†). The ESR spectrum of 1˙+ in CH2Cl2 at 298 K showed a five-line splitting, which is mainly attributed to the hyperfine coupling interactions from two equivalent nitrogen nuclei (|aN| = 0.58 mT) (Fig. 6). This result indicates that the generated radical spin is equally delocalized over the two phenothiazine moieties. Oxidation of 1 with more than 2 equiv. of magic blue did not give the crystals of 12+[SbCl6−]2, because of the high second oxidation potential of 1 comparable to the formal potential of magic blue.23 For 2, the chemical oxidation with magic blue gave the crystals of doubly oxidized species, 22+[SbCl6−]2, even when treated with 1 equiv. of oxidant because of the disproportionation reaction. The structure of 22+ was also shown by the X-ray analysis (Fig. S14, ESI†). The dication 22+ maintained a cruciform orientation, and both the two phenothiazine moieties took more planar structure compared to the neutral 2.
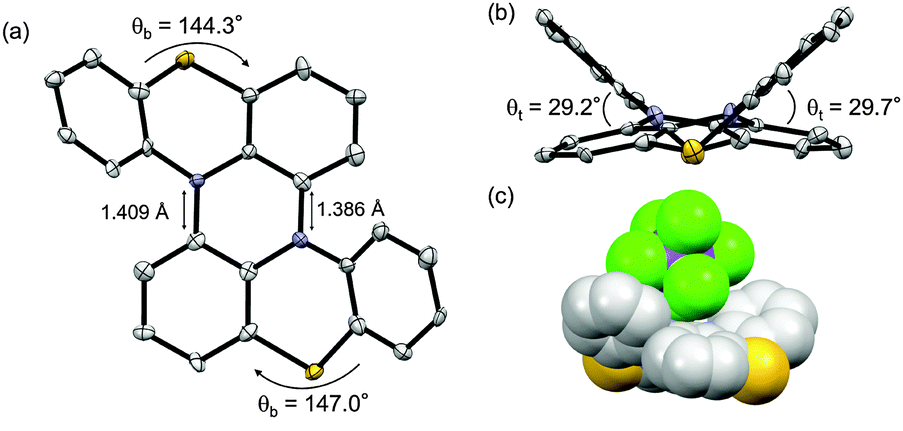 | ||
| Fig. 5 X-ray structure of 1˙+SbCl6−. (a) Top view and (b) side view of 1˙+. (c) Space-filling representation of 1˙+SbCl6−. Thermal ellipsoids are set at 50% probability. The tBu groups and hydrogen atoms were omitted for clarity. | ||
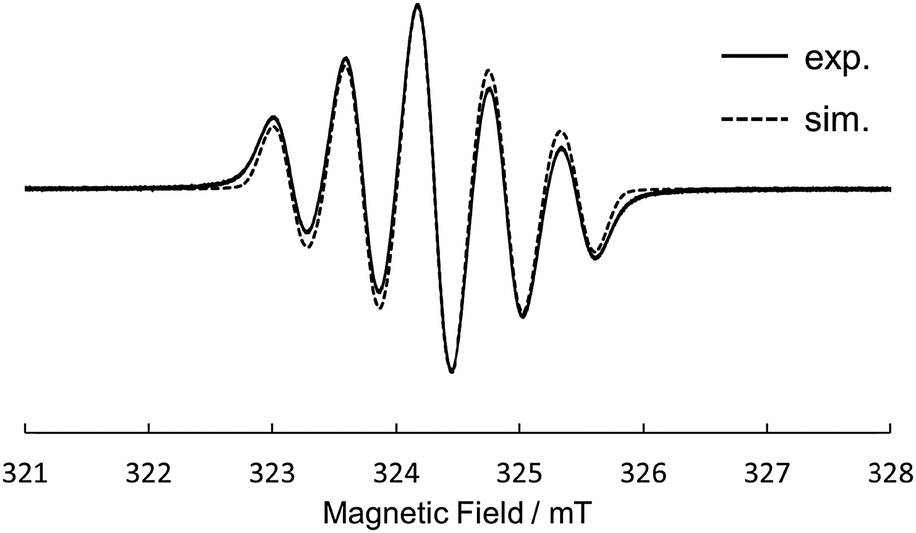 | ||
| Fig. 6 ESR spectrum of 1˙+ (g = 2.0038) in CH2Cl2 at 298 K. | ||
In summary, we have synthesized for the first time the doubly fused phenothiazine dimer 1 with a double hetero[4]helicene structure by using the tandem oxidation of the phenothiazine derivative via the singly connected dimer 2. Although double[4]helicenes usually racemize quickly, the enantiomers of 1 were much more stable toward racemization stemming from the highly bent structure of phenothiazine. Both 1 and 2 exhibited good electron donor properties, and the chemical oxidation of these dimers gave the radical cations stable enough to be isolated. X-ray single-crystal analysis clarified the structures of 1˙+ and 22+. The torsion angles in the fjord regions of 1˙+ were found to be decreased compared with those in the neutral state. The generated spin of 1˙+ was fully delocalized over the double[4]helicene skeleton. Further studies on analogues of 1 with other heteroatoms are currently underway.
We are deeply thankful to Dr Nobuko Kanehisa (Osaka University) for her help with single-crystal X-ray analyses. This work was partly supported by a Grant-in-Aid for Young Scientists (B) (26810023) from the Japan Society for the Promotion of Science (JSPS) and by the Nanotechnology Platform Program (Molecule and Material Synthesis) of the Ministry of Education, Culture, Sports, Science and Technology, Japan. D.S. thanks the JSPS Research Fellowship for Young Scientists. The theoretical calculations were performed using the Supercomputer System of Kyoto University and the Research Center for Computational Science in Okazaki (Japan).
Notes and references
- (a) K. Okada, T. Imakura, M. Oda, H. Murai and M. Baumgarten, J. Am. Chem. Soc., 1996, 118, 3047 CrossRef CAS; (b) K. Okada, T. Imakura, M. Oda, A. Kajiwara, M. Kamachi, K. Sato, D. Shiomi, T. Takui, K. Itoh, L. Gherghel and M. Baumgarten, J. Chem. Soc., Perkin Trans. 2, 1997, 1059 RSC; (c) E. Terada, T. Okamoto, M. Kozaki, M. E. Masaki, D. Shiomi, K. Sato, T. Takui and K. Okada, J. Org. Chem., 2005, 70, 10073 CrossRef CAS PubMed.
- D. Sun, S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2004, 126, 1388 CrossRef CAS PubMed.
- S. Suzuki, K. Yoshida, M. Kozaki and K. Okada, Angew. Chem., Int. Ed., 2013, 52, 2499 CrossRef CAS PubMed.
- A. C. Jahnke, M. Spulber, M. Neuburger, C. G. Palivan and O. S. Wenger, Chem. Commun., 2014, 50, 10883 RSC.
- Y. Tsujino, Tetrahedron Lett., 1968, 9, 4111 CrossRef.
- G. Cauquis, A. Deronzier and D. Serve, J. Electroanal. Chem., 1973, 47, 193 CrossRef CAS.
- C. S. Krämer, K. Zeitler and T. J. J. Müller, Tetrahedron Lett., 2001, 42, 8619 CrossRef.
- T. Okamoto, M. Kuratsu, M. Kozaki, K. Hirotsu, A. Ichimura, T. Matsushita and K. Okada, Org. Lett., 2004, 6, 3493 CrossRef CAS PubMed.
- D.-M. S. Islam, Y. Sasaki, H. Kawauchi, M. Kozaki, Y. Araki, O. Ito and K. Okada, Bull. Chem. Soc. Jpn., 2008, 81, 103 CrossRef CAS.
- A. W. Franz, F. Rominger and T. J. J. Müller, J. Org. Chem., 2008, 73, 1795 CrossRef CAS PubMed.
- H. Oka, Org. Lett., 2010, 12, 448 CrossRef CAS PubMed.
- H. Kanazawa, M. Higuchi and K. Yamamoto, J. Am. Chem. Soc., 2005, 127, 16404 CrossRef CAS PubMed.
- Recent reports of the synthesis of double helicenes, see (a) K. Shiraishi, A. Rajca, M. Pink and S. Rajca, J. Am. Chem. Soc., 2005, 127, 9312 CrossRef CAS PubMed; (b) Z. Wang, J. Shi, J. Wang, C. Li, X. Tian, Y. Cheng and H. Wang, Org. Lett., 2010, 12, 456 CrossRef CAS PubMed; (c) J. Luo, X. Xu, R. Mao and Q. Miao, J. Am. Chem. Soc., 2012, 134, 13796 CrossRef CAS PubMed; (d) X. Liu, P. Yu, L. Xu, J. Yang, J. Shi, Z. Wang, Y. Cheng and H. Wang, J. Org. Chem., 2013, 78, 6316 CrossRef CAS PubMed; (e) K. Nakamura, S. Furumi, M. Takeuchi, T. Shibuya and K. Tanaka, J. Am. Chem. Soc., 2014, 136, 5555 CrossRef CAS PubMed; (f) S. Hashimoto, S. Nakatsuka, M. Nakamura and T. Hatakeyama, Angew. Chem., Int. Ed., 2014, 53, 14074 CrossRef CAS PubMed; (g) H. Kashihara, T. Asada and K. Kamikawa, Chem. – Eur. J., 2015, 21, 6523 CrossRef CAS PubMed; (h) T. Fujikawa, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 7763 CrossRef CAS PubMed.
- D. Sakamaki, D. Kumano, E. Yashima and S. Seki, Angew. Chem., Int. Ed., 2015, 54, 5404 CrossRef CAS PubMed.
- R. Kuroda, J. Chem. Soc., Perkin Trans. 2, 1982, 789 RSC.
- All the DFT calculations in this study were carried out by using Gaussian 09 program package (Revision C.01).
- M. Kuratsu, M. Kozaki and K. Okada, Chem. Lett., 2004, 33, 1174 CrossRef CAS.
- (a) G. Lamanna, C. Faggi, F. Gasparrini, A. Ciogli, C. Villani, P. J. Stephens, F. J. Devlin and S. Menichetti, Chem. – Eur. J., 2008, 14, 5747 CrossRef CAS PubMed; (b) S. Menichetti, S. Cecchi, P. Procacci, M. Innocenti, L. Becucci, L. Franco and C. Viglianisi, Chem. Commun., 2015, 51, 11452 RSC.
- R. H. Janke, G. Haufe, E.-U. Würthwein and J. H. Borkent, J. Am. Chem. Soc., 1996, 118, 6031 CrossRef CAS.
- Examples of the redox-active heterohelicenes, see (a) A. Rajca, M. Miyasaka, M. Pink, H. Wang and S. Rajca, J. Am. Chem. Soc., 2004, 126, 15211 CrossRef CAS PubMed; (b) J. K. Zak, M. Miyasaka, S. Rajca, M. Lapkowski and A. Rajca, J. Am. Chem. Soc., 2010, 132, 3246 CrossRef CAS PubMed; (c) K. Goto, R. Yamaguchi, S. Hiroto, H. Ueno, T. Kawai and H. Shinokubo, Angew. Chem., Int. Ed., 2012, 51, 10333 CrossRef CAS PubMed.
- D. W. Cho, M. Fujitsuka, A. Sugimoto, U. C. Yoon, P. S. Mariano and T. Majima, J. Phys. Chem. B, 2006, 110, 11062 CrossRef CAS PubMed.
- A. Ito, D. Sakamaki, Y. Ichikawa and K. Tanaka, Chem. Mater., 2011, 23, 841 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, and spectroscopic and computational data. CCDC 1415094–1415097. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc07680c |
| This journal is © The Royal Society of Chemistry 2015 |