
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and luminescence modulation of pyrazine-based gold(III) pincer complexes†
Julio
Fernandez-Cestau
a,
Benoît
Bertrand
a,
Maria
Blaya
a,
Garth A.
Jones
a,
Thomas J.
Penfold
*b and
Manfred
Bochmann
*a
aSchool of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: m.bochmann@uea.ac.uk
bDepartment of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: Tom.Penfold@newcastle.ac.uk
First published on 25th September 2015
Abstract
The first examples of pyrazine-based gold(III) pincer complexes are reported; their intense photoemissions can be modified by protonation, N-alkylation or metal ions, without the need for altering the ligand framework. Emissions shift from red (77 K) to blue (298 K) due to thermally activated delayed fluorescence (TADF).
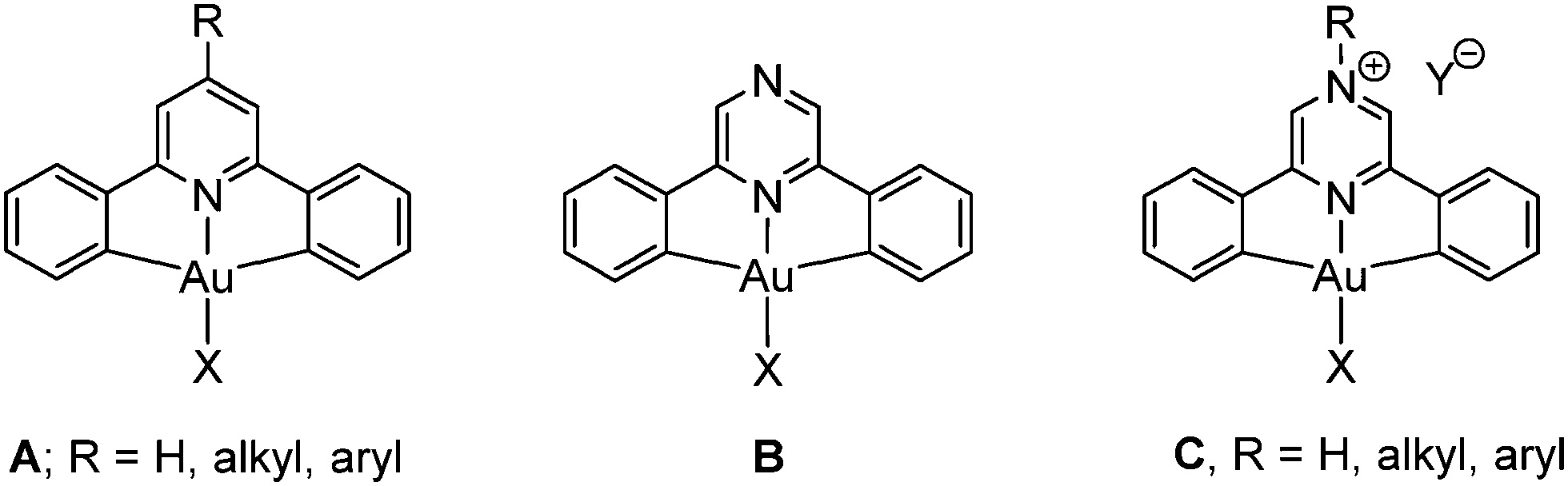
Gold(III) complexes with bis-cyclometallated ligands are characterized by their chemical stability and resistance to reduction. The ligand scaffold based on 2,6-diphenylpyridine1 has proved particularly useful in gold(III) chemistry and forms (C^N^C)AuX pincer complexes of type A (Chart 1). Such complexes have proved to be highly versatile and, in combination with strong carbon-based σ-donor ligands (e.g. X = N-heterocyclic carbene, alkynyl), display interesting photophysical2–4 properties. This C^Npy^C ligand system has also been successful in stabilizing types of compounds that have frequently been invoked as unstable intermediates in catalytic cycles or postulated in computer modelling of catalytic processes, such as gold(III) hydrido, alkene, CO and peroxo complexes.5
 | ||
| Chart 1 | ||
Photoemissive materials as components of electronic devices such as flat screen displays should ideally be capable of covering the whole range of the visible spectrum. Such changes in emission colours can be induced by suitable modification of the ligand framework. For (C^N^C)Au(III) complexes a widely applied strategy for modulating the photoluminescence (PL) response and widening the range of emission wavelengths has been the introduction of electron donating or withdrawing substituents in the 4-position of the pyridine moiety.3,4 There are however limitations in this approach: firstly, the synthesis of C^N^C gold complexes involves two C–H activation steps, each of which is sensitive to the ligand structure and needs to be optimized for each new ligand; secondly, the modulation of electronic characteristics of the central pyridine moiety that can be achieved by inductive or mesomeric substituent effects is limited.
Rather more profound electronic changes in C^N^C ligands can be introduced by replacing the central pyridine ring by other heterocycles, such as pyrazine, to give compounds of type B. The lowest-energy π–π* transition in pyrazine is about 0.95 eV smaller than in pyridine;6,7 pyrazine ligands are therefore much better electron acceptors and are likely to produce a red-shift of their UV and photoemission wavelengths. In addition, pyrazine complexes B offer scope for further derivatisation by protonation or quaternisation of the non-coordinating N atom, to give salts of type C.
Complexes of aryl-substituted pyrazines and quinoxalines are of course well-known for iridium(III) and platinum(II), since for these metals they are readily accessible by direct cyclometallation of the neutral ligand precursors by noble metal halides.8 By contrast, related gold(III) complexes have until now been inaccessible since the usual methods employed for the synthesis of pyridine complexes A fail for the analogous pyrazine derivatives. Here we report the first examples of cyclometallated Au(III) pyrazine complexes and the facile modification of their photoluminescence properties.
The mercuration of the pro-ligand 2,6-bis(4′-t-BuC6H4)2pz (pz = pyrazine) requires forcing conditions but proceeds using Hg(tfa)2 in Htfa (tfa = CF3CO2) to give (C^Npz^C)HgCl·2Htfa (1). Transmetallation with KAuCl4 affords (C^Npz^C)AuCl (2) as a yellow crystalline powder in good yield (Scheme 1).
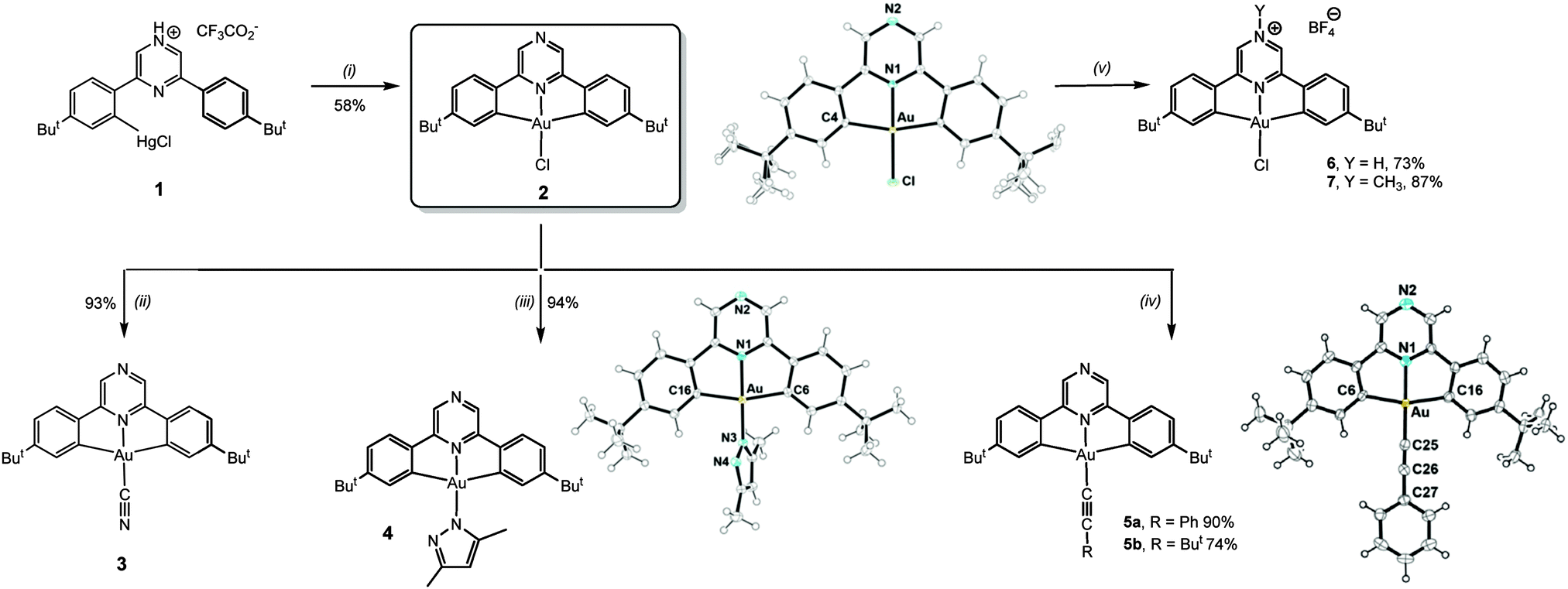 | ||
Scheme 1 Conditions: (i) KAuCl4, CH3CN/H2O 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, reflux 72 h. (ii) KCN, CH2Cl2, 20 °C, 24 h. (iii) KOtBu, 3,5-dimethylpyrazole, toluene, 20 °C, 6 h. (iv) AgC :1, reflux 72 h. (ii) KCN, CH2Cl2, 20 °C, 24 h. (iii) KOtBu, 3,5-dimethylpyrazole, toluene, 20 °C, 6 h. (iv) AgC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR, CH2Cl2, 20 °C, 24 h. (v) HBF4·Et2O, Et2O, 20 °C, 30 min. (6), or [Me3O]BF4, toluene/1,2-C6H4F2 (5:1 v/v), 12 h. (7). Molecular structures [selected bond distances (Å) and angles (°)]: 2: Au–N1 1.972(3), Au–Cl 2.2651(11), Au–C4 2.078; N1–Au–C4 81.28(8), C4–Au–Cl 98.72(8), C4–Au–C4′ 162.56(16), N1–Au–Cl 180. 4: Au–N1 1.979(2), Au–C6 2.080(3), Au–C16 2.073(3), Au–N3 1.991(2); C6–Au–N1 81.39(10), C16–Au–N1 81.32(11), C6–Au–N3 98.88(10), C16–Au–N3 98.38(11), N1–Au–N3 178.33(9). 5a: Au–N1 1.997(5), Au–C6 2.070(6), Au–C16 2.083(7), Au–C25 1.979(7), C25–C26 1.199(9); N1–Au–C6 80.4(2), N1–Au–C16 81.2(2), C6–Au–C25 99.3(3), C16–Au–C25 99.0(3), C6–Au–C16 161.6(3), Au–C25–C26 175.9(6), C25–C26–C27 177.4(7). CR, CH2Cl2, 20 °C, 24 h. (v) HBF4·Et2O, Et2O, 20 °C, 30 min. (6), or [Me3O]BF4, toluene/1,2-C6H4F2 (5:1 v/v), 12 h. (7). Molecular structures [selected bond distances (Å) and angles (°)]: 2: Au–N1 1.972(3), Au–Cl 2.2651(11), Au–C4 2.078; N1–Au–C4 81.28(8), C4–Au–Cl 98.72(8), C4–Au–C4′ 162.56(16), N1–Au–Cl 180. 4: Au–N1 1.979(2), Au–C6 2.080(3), Au–C16 2.073(3), Au–N3 1.991(2); C6–Au–N1 81.39(10), C16–Au–N1 81.32(11), C6–Au–N3 98.88(10), C16–Au–N3 98.38(11), N1–Au–N3 178.33(9). 5a: Au–N1 1.997(5), Au–C6 2.070(6), Au–C16 2.083(7), Au–C25 1.979(7), C25–C26 1.199(9); N1–Au–C6 80.4(2), N1–Au–C16 81.2(2), C6–Au–C25 99.3(3), C16–Au–C25 99.0(3), C6–Au–C16 161.6(3), Au–C25–C26 175.9(6), C25–C26–C27 177.4(7). | ||
Substitution of the chloride ligand in 2 with KCN gives the cyanide complex 3, while treatment with dimethylpyrazole affords the pyrazolato complex 4. The reaction with AgCCR in CH2Cl2 affords the acetylides (C^Npz^C)AuCCR (5a, R = Ph; 5b, R = But) in essentially quantitative yield.
Pyrazine is only weakly basic (pKa 1.30, vs. 5.20 of pyridine).9 However, protonation of 2 by HBF4·OEt2 generated the corresponding salt 6. Remarkably, the protonation by dry HCl in Et2O proved to be reversible, and evaporation of the solvent from the HCl adduct regenerated neutral 2. The alkylation of the non-coordinating pyrazine N-atom was achieved using Meerwein's salt [Me3O]BF4 to give 7 (Scheme 1) as a deep-red crystalline solid in 87% yield. The molecular structures10 of 2, 4, 5a and 5b were determined by X-ray diffraction (ESI†). The interatomic distances and angles are as expected for square-planar pincer complexes of this type (see ESI† for details).
All the complexes show moderate to intense luminescence in the solid state and in solution (for a detailed summary see the ESI,† Tables S2 and S3). Both the UV-vis spectra and the PL bands are characterized by the vibronic progression of the C^Npz^C pincer. As expected on the basis of the lower π–π* energy gap of the pyrazine compounds, the bands are red-shifted by about 40–50 nm compared with their C^Npy^C analogues (see Fig. S22, ESI†).
Whereas in the case of diphenylpyridine-based complexes of type A strongly σ-donating ligands (such as aryls, acetylides or N-heterocyclic carbenes3,4) are required to raise the d-orbital energy of Au(III) and increase the 3LLCT contribution to the emissive state, the pyrazine derivatives 2–7 reported here are emissive even if ligands X are weaker heteroatom donors. This is best exemplified by the chlorides: whereas (C^Npy^C)AuCl is non-emissive at room temperature,1 complex 2 shows noticeable naked eye emission in fluid media (CH2Cl2 or 2-MeTHF) at 298 K that becomes very intense at 77 K (Fig. S20, ESI†). Similarly, the pyrazolato complex 4 is brightly emissive in the solid state or in fluid media at 298 K, while under identical conditions, the C^Npy^C analogue is only weakly emissive.11 The pyrazine ligand framework of type B therefore significantly widens the range of photoemissive metal–ligand combinations.
In solution, the neutral complexes 2–5 all emit in the yellow to green region of the spectrum, mainly due to a triplet state based on the diphenylpyrazine pincer ligand. The excited state lifetimes show biexponential decay, with the fast component in the range of 5–20 ns.
At 77 K the emissions of 2, both in the solid state (λmaxem 563 nm) and in solution (λmaxem = 532 nm), agree reasonably well with the value calculated for a T1 → S0 transition (λcalcem = 541 nm).‡ However, at 298 K the emission shows a significant blue-shift, to 482 nm, a feature that is even more pronounced on addition of acid (vide infra).
One of the most important challenges in the design of photoluminescent devices is the ability to modulate the energy of the emitted light. In the pyrazine ligand system, the easiest way to achieve this is by making use of the non-coordinating nitrogen of the pyrazine ring. Strong Brønsted and Lewis acids do indeed produce dramatic changes in PL response. In order to eliminate any possible anion effects, initial protonation studies were carried out using the solid Brønsted acid [H(OEt2)2][H2N{B(C6F5)3}2]12 (“HNB2”). As shown in Fig. 1a, protonating 2 with this acid results in a blue-shift compared with the neutral complex, from 482 to 458 nm. On the other hand, at 77 K the emissions of both the neutral and protonated compounds are remarkable similar (as expected since both the HOMO and LUMO are based on the pyrazine ligand and are equally affected by protonation) and the emission is due to an intra-ligand charge transfer (3ILCT) process.
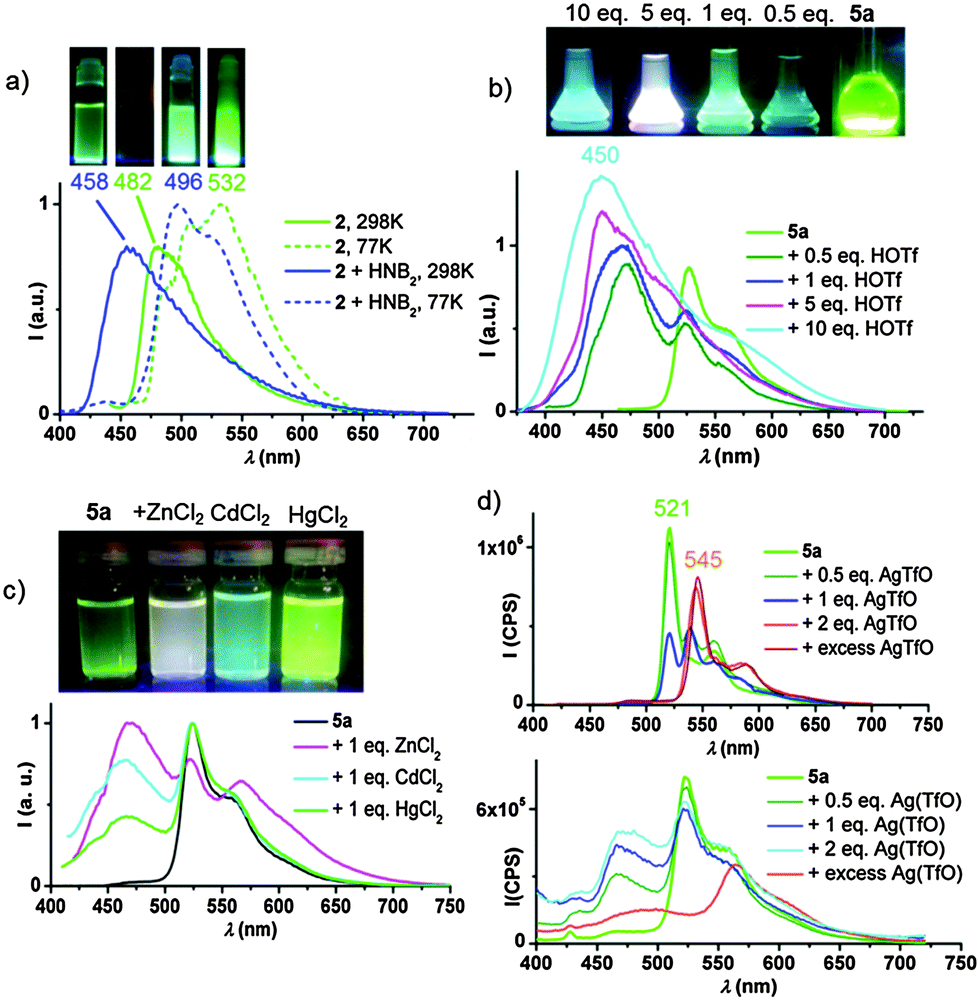 | ||
| Fig. 1 (a) PL of 2 (CH2Cl2, 1 × 10−4 M) in the absence and presence of two equivalents of [H(OEt2)2][H2N{B(C6F5)3}2] (HNB2) at 298 and 77 K. (b) PL response of 5a (CH2Cl2, 1 × 10−4 M) to the addition of HOTf at 298 K. (c) PL of mixtures of 5a with equimolar amounts of MCl2 (M = Zn, Cd, Hg) in THFMe-2 ([5a] = 10−4 M) at 298 K. (d) PL response of 5a (THFMe-2, 5 × 10−4 M) to the addition of AgTfO at 77 (top) and 298 K (bottom). | ||
The temperature-dependence of the photoluminescence response is in agreement with a TADF process,13 where the energy difference between the T1 and S1 excited states is sufficiently small to allow thermally driven repopulation of the S1 state from the T1 state, with the faster decay occurring via the S1 → S0 transition. In agreement with this mechanism, the quantum yields for the protonated pyrazine systems are about an order of magnitude higher than those of the neutral complexes. Photoemissions via a TADF mechanism are common for copper(I);14 but have not been previously reported for Au(III).
The alkynyl complexes 5a and 5b show similar behaviour: the gradual addition of triflic acid results in the progressive growth of a blue emission (λmaxem = 450 nm for 5a, Fig. 1b). Emissions involve a mixture of states, with both blue and green components, which combine to appear white.
The same effect can be achieved with the Lewis acid B(C6F5)3. In this case the high energy component appears at slightly lower energy (λmaxem = 481 nm), to give a more pronounced blue effect.
Whereas in the case of the chloride 2 both HOMO and LUMO were based on the pincer ligand, for 5a the excitation involves metal-perturbed ligand-to-ligand π(CCPh) → π*(C^Npz^C) charge transfer (3LLCT, also involving HOMO−2). The effect of adding H+ or B(C6F5)3 to the pyrazine N-atom is therefore pronounced. DFT calculations predict a strong red shift on forming the 5a-H+ cation since on protonation the HOMO–LUMO gap is reduced (e.g. calculated HOMO–LUMO difference for 5a = 5.963 eV, compared to 4.878 eV after H+ addition). In agreement with this, solutions of 5a·B(C6F5)3 at 77 K display an intense emission at 569 nm (Fig. S26, ESI†) which is gradually deactivated on warming, while at the same time a high energy component increases, resulting in the blue emission at λmaxem = 481 nm at 298 K. As can be seen (Fig. S29, ESI†), these reactions also occur in the solid state.
Different metal ions also lead to distinctive modulations of the PL emission, arising from a mixture of states. The addition of ZnCl2, CdCl2 and HgCl2 (1:1 molar ratio) results in white, turquoise and light green emissions, respectively (Fig. 1c). The gradual addition of ZnCl2 to a solution of the complex 5a in 2-MeTHF, results in the activation of the blue TADF component at the expense of the green phosphorescence (see Fig. S27, ESI†). Cooling to 77 K recovers the phosphorescent emission.
The addition of AgOTf or CuOTf also enhances the emission via the TADF mechanism (see Fig. 1d for Ag+ and Fig. S28, ESI† for Cu+). Freezing 2-MeTHF solutions of 5a/AgOTf to 77 K reveals the presence of two different systems; complex 5a (λmaxem = 521 nm), and a second complex characterized by a band with maximum at 545 nm. The mixture of 5a with two equivalents and with an excess of Ag+ both show the same emission band, while 1 equivalent of Ag+ gives two bands of about equal intensity. For these reasons, we tentatively suggest the formation of an aggregate of 5a with two Ag+ ions (e.g. coordination of two Ag+ ions to the CC bond and the pyrazine-N atom may be envisaged).
The N-methylated complex 7 mirrors the behaviour of the H+ adducts and shows blue emission in solution (λmaxem = 460 nm) but is dark-red in the solid state (λem = 623max, 680sh).
In summary, cyclometallated gold(III) pincer complexes based on pyrazine provide a new family of photoluminescent compounds which allow facile modulation of the emission characteristics by protonation, alkylation, Lewis acids or metal ions, without the need for modifying the pincer ligand framework. The modulation arises from the coexistence of high energy TADF and 3IL (C^Npz^C)/3LLCT (X → C^Npz^C) transitions.
This work was supported by the European Research Council. M. B. is an ERC Advanced Investigator Award holder (grant no. 338944-GOCAT).
Notes and references
- K.-H. Wong, K.-K. Cheung, M. C.-W. Chan and C.-M. Che, Organometallics, 1998, 17, 3505 CrossRef CAS.
- Reviews: (a) C. Bronner and O. S. Wenger, Dalton Trans., 2011, 40, 12409 RSC; (b) V. W.-W. Yam and K. M. C. Wong, Chem. Commun., 2011, 47, 11579 RSC.
- (a) V. K.-M. Au, D. P.-K. Tsang, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, J. Organomet. Chem., 2015, 792, 109 CrossRef CAS; (b) M.-C. Tang, D. P.-K. Tsang, Y.-C. Wong, M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2014, 136, 17861 CrossRef CAS PubMed and cited refs.
- (a) G. Cheng, K. T. Chan, W.-P. To and C.-M. Che, Adv. Mater., 2014, 26, 2540 CrossRef CAS PubMed; (b) W.-P. To, K. T. Chan, G. S. M. Tong, C. S. Ma, W.-M. Kwok, X. G. Guan, K. H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 6648 CrossRef CAS PubMed; (c) W.-P. To, G. S.-M. Tong, W. Lu, C.-S. Ma, J. Liu, A. L.-F. Chow and C.-M. Che, Angew. Chem., Int. Ed., 2012, 51, 2654 CrossRef CAS PubMed.
- (a) D.-A. Roşca, D. A. Smith, D. L. Hughes and M. Bochmann, Angew. Chem., Int. Ed., 2012, 51, 10643 CrossRef PubMed; (b) N. Savjani, D.-A. Roşca, M. Schormann and M. Bochmann, Angew. Chem., Int. Ed., 2013, 52, 874 CrossRef CAS PubMed; (c) D.-A. Roşca, J. A. Wright, D. L. Hughes and M. Bochmann, Nat. Commun., 2013, 4, 2167 Search PubMed; (d) D.-A. Roşca, J. Fernandez-Cestau, J. Morris, J. A. Wright and M. Bochmann, Sci. Adv., 2015 Search PubMed , in press.
- C. Walker, M. H. Palmer and A. Hopkirk, Chem. Phys., 1989, 141, 365 CrossRef.
- I. C. Walker and M. H. Palmer, Chem. Phys., 1991, 153, 169 CrossRef CAS.
- See for example: (a) S. H. Wu, S. E. Burkhardt, J. Yao, Y. W. Zhong and H. D. Abruña, Inorg. Chem., 2011, 50, 3959 CrossRef CAS PubMed; (b) V. N. Kozhevnikov, M. C. Durrant and J. A. G. Williams, Inorg. Chem., 2011, 50, 6304 CrossRef CAS PubMed; (c) S. Culham, P.-H. Lanoë, V. L. Whittle, M. C. Durrant, J. A. G. Williams and V. N. Kozhevnikov, Inorg. Chem., 2013, 52, 10992 CrossRef CAS PubMed; (d) P.-H. Lanë, C.-M. Tong, R. W. Harrington, M. R. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Chem. Commun., 2014, 50, 6831 RSC.
- H. J. Soscún Machado and A. Hinchliffe, THEOCHEM, 1995, 339, 255 CrossRef.
- S. J. Coles and P. Gale, Chem. Sci., 2012, 3, 683 RSC.
- D.-A. Roşca, D. A. Smith and M. Bochmann, Chem. Commun., 2012, 48, 7247 RSC.
- S. J. Lancaster, A. Rodriguez, A. Lara-Sanchez, M. D. Hannant, D. A. Walker, D. L. Hughes and M. Bochmann, Organometallics, 2002, 21, 451 CrossRef CAS.
- Reviews: (a) H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622 CrossRef CAS; (b) Y. Tao, K. Yuan, T. Chen, P. Xu, H.-H. Li, R. F. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931 CrossRef CAS PubMed.
- (a) M. J. Leitl, V. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032 CrossRef CAS PubMed; (b) V. A. Krylova, P. I. Djurovich, B. L. Conley, R. Haiges, M. T. Whited, T. J. Williams and M. E. Thompson, Chem. Commun., 2014, 50, 7176 RSC; (c) R. Marion, F. Sguerra, F. Di Meo, E. Sauvageot, J.-F. Lohier, R. Daniellou, J.-L. Renaud, M. Linares, M. Hamel and S. Gaillard, Inorg. Chem., 2014, 53, 9181 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of synthesis and characterization, X-ray crystallography, photophysical properties, theoretical calculations. CCDC 1417819–1417822. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc07523h |
| ‡ In the solid state at room temperature, complex 2 is non-emissive, most probably due to close intermolecular interactions in the crystal lattice which quench the luminescence. This notion is supported by the observed higher solid-state emission intensity of 5b (λmax = 523 nm, φ = 8.3) compared to 5a (λmax = 523 nm, φ = 4.5); the crystal packing shows that for 5b intermolecular π-stacking is disfavoured on steric grounds. In solution at 298 K the PL intensities of 5a (λmax = 526 nm, φ = 0.462) and 5b (λmax = 526 nm, φ = 0.512) are essentially identical (see Fig. S19, ESI†). |
| This journal is © The Royal Society of Chemistry 2015 |