
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereospecific SN2@P reactions: novel access to bulky P-stereogenic ligands†‡
Sílvia
Orgué
a,
Areli
Flores-Gaspar§
a,
Maria
Biosca
b,
Oscar
Pàmies
b,
Montserrat
Diéguez
b,
Antoni
Riera
*ac and
Xavier
Verdaguer
*ac
aInstitute for Research in Biomedicine (IRB Barcelona), The Barcelona Institute of Science and Technology, Baldiri Reixac 10, E-08028, Spain. E-mail: antoni.riera@irbbarcelona.org; xavier.verdaguer@irbbarcelona.org; Fax: +34-934037095; Tel: +34-934034813
bDepartment Química Física i Inorgànica, Universitat Rovira i Virgili, Marcel lí Domingo s/n, 43007 Tarragona, Spain
cDepartment de Química Orgànica, Universitat de Barcelona, Martí i Franquès 1, E-08028 Barcelona, Spain
First published on 2nd October 2015
Abstract
The stereospecific hydrolysis of bulky aminophosphine boranes is reported for the first time. The resulting phosphinous acid boranes, upon activation, undergo stereospecific nucleophilic substitution reaction at the phosphorous center with amine nucleophiles. The combination of these two processes provides a novel access to bulky P*-ligands.
Chiral phosphines are the cornerstone of asymmetric metal catalysis.1 Among this class of ligands, bulky P-stereogenic phosphines have proved to be highly efficient in asymmetric hydrogenation and other relevant processes.2,3 However, their synthesis in the optically pure form is often not straightforward. The stereoselective synthesis of tert-butyl P-stereogenic phosphines has relied mostly on the selective deprotonation of tert-butyldimethylphosphine borane complexes4 and on the reaction of borane lithium phosphides with electrophiles.5 Nucleophilic substitution reactions at the “bulky” P-center (SN2@P) are generally avoided since it is overly unreactive. Furthermore, when forced to react, it usually provides non-stereospecific substitution processes.6,7 A relevant exception to this behavior is the reaction of halo-tert-butylmethylphosphine-boranes with alkynyllithium reagents.8 However, halophosphines are configurationally unstable and have to be generated and reacted in situ at low temperature.
Jugé and others showed that P-stereogenic aryl and alkyl aminophosphine boranes undergo stereospecific acid-promoted methanolysis to yield the resulting methyl phosphinites with inversion at the P-center.9 The acidolysis can also be carried out with HCl/toluene, in this case yielding the optically enriched chlorophosphine boranes.10 However, these reactions directly fail or provide reduced optical purity when a bulky group (e.g., tert-butyl) is attached to phosphorus.
We recently reported the stereospecific synthesis of aminophosphines 1a and 1b which have been used by us in the preparation of MaxPHOS and SIP type ligands.11 We considered that these compounds were ideally suited to evaluate whether the acidolysis of bulky aminophosphines could be performed in a stereospecific manner, and whether the resulting products could be further transformed into valuable ligand structures (Scheme 1). Herein we report on the stereospecific hydrolysis of bulky aminophosphine boranes and how the resulting phosphinous acids can be transformed into P*-ligands through SN2@P reactions.
 | ||
| Scheme 1 Novel stereospecific route to bulky P-stereogenic phosphines. | ||
The initial acidolysis studies on 1a and 1b were conducted in H2SO4/MeOH (Table 1). Methanolysis of 1a at 50 °C for 16 h did not afford the expected methyl phosphinite borane 2a but the corresponding phosphinous acid borane 3a and the secondary phosphine oxide 4a that results from borane deprotection and P(III)/P(V) tautomerization of 3a (Table 1, entry 1). We attributed the formation of 3a to the residual content of water in the solvent used. To confirm this hypothesis, we next ran the acidolysis reaction of 1a in a MeOH/H2O (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture. This solvent mixture and heating to 40 °C for 16 h afforded exclusively phosphine oxide 4a in low enantiomeric excess (Table 1, entry 2). By simply reducing the reaction time to 2 h, we were able to isolate the corresponding phosphinous acid 3a with an excellent yield and optical purity (>99% ee, Table 2, entry 3). The hydrolysis of 1b was also carried out stereospecifically to yield the corresponding phosphinous acid 3b in >99% ee and 63% yield (Table 1, entry 4). Hydrolysis of 1a and 1b took place with inversion of the configuration at the P-center as confirmed by X-ray crystallography of the corresponding benzoyl derivative (see ESI‡).12 The opposite enantiomer of 3a and and 3b were obtained when starting from (RP)-1a and (SP)-1b thus confirming that the process is completly stereospecific. These results indicate that the acidolysis of bulky aminophosphines is extremely sensitive to the nature of the incoming nucleophile. We believe that the minimal steric differences between methanol and water allow the latter to act as an efficient nucleophile, while making the former unreactive.
:1) mixture. This solvent mixture and heating to 40 °C for 16 h afforded exclusively phosphine oxide 4a in low enantiomeric excess (Table 1, entry 2). By simply reducing the reaction time to 2 h, we were able to isolate the corresponding phosphinous acid 3a with an excellent yield and optical purity (>99% ee, Table 2, entry 3). The hydrolysis of 1b was also carried out stereospecifically to yield the corresponding phosphinous acid 3b in >99% ee and 63% yield (Table 1, entry 4). Hydrolysis of 1a and 1b took place with inversion of the configuration at the P-center as confirmed by X-ray crystallography of the corresponding benzoyl derivative (see ESI‡).12 The opposite enantiomer of 3a and and 3b were obtained when starting from (RP)-1a and (SP)-1b thus confirming that the process is completly stereospecific. These results indicate that the acidolysis of bulky aminophosphines is extremely sensitive to the nature of the incoming nucleophile. We believe that the minimal steric differences between methanol and water allow the latter to act as an efficient nucleophile, while making the former unreactive.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Conditionsa | 2a/2b | 3a/3bb | 4a/4b |
| a Aminophosphine (7.5 mmol), H2SO4 (30 mmol) and 42 mL of solvent were used. b Isolated yield after flash column chromatography. c Conversion data based on 1H NMR crude reaction. d Enantiomeric excess determined by chiral GC or HPLC analysis of the corresponding methylated derivative 2a/2b. e Enantiomeric excess determined by optical rotation. | |||||
| 1 | Me | MeOH 50 °C, 16 h | — | 75%c | 25%c |
| 2 | Me | MeOH/H2O (20:1) 40 °C, 16 h |
— | — | 83% (53% ee)e |
| 3 | Me | MeOH/H2O (20:1) 50 °C, 2 h |
— | 97% (>99% ee)d | — |
| 4 | Ph | MeOH/H2O (2:1) 50 °C, 7 h |
— | 63% (>99% ee)d | — |

|
|
|||||
|---|---|---|---|---|---|
| Entry | SM | Nucleophile | Product | Yielda (%) | ee/deb (%) |
| a Isolated yield after flash column chromatography. b Enantiomeric excess determined by either chiral GC or HPLC analysis, diastereomeric excess determined by 1H NMR of the crude reaction. c Optical purity of 7 was assigned tentatively by analogy with similar primary amines tested. d Determination of optical purity by chromatographic methods failed. [α]D = −23.5° (c 1.00, CHCl3). e Determination of optical purity by chromatographic methods failed. [α]D = −2.1° (c 1.49, CHCl3). nd = not determined. | |||||
| 1 | 3a | NH3 | (RP)-1a | 99 | 96 ee |
| 2 | 3a |
|
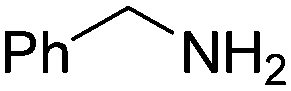
(RP)-6 | 60 | 98 ee |
| 3 | 3a |

|
(RP)-7c | 64 | >95 ee |
| 4 | 3a |
|
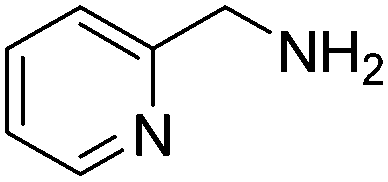
(RP)-8 | 87 | 99 ee |
| 5 | 3a |

|
(RP)-9 | 73 | 96 ee |
| 6 | 3a |

|
(RP)-10 | 76 | 98 de |
| 7 | 3a |

|
(RP)-11 | 71 | 95 de |
| 8 | 3a |

|
(RP)-12 | 63 | 97 de |
| 9 | 3a |

|
(RP)-13 | 65 | 98 ee |
| 10 | 3a |

|
(RP)-14 | 42 | 91 ee |
| 11 | 3a | HNBn2 | (RP)-15 | 0 | — |
| 12 | 3a |

|
(RP)-16 | 0 | — |
| 13 | 3a |
|
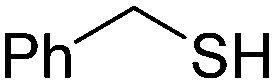
(RP)-17d | 22 | nd |
| 14 | 3a | PhSH | (RP)-18e | 36 | nd |
| 15 | 3b | NH3 | (SP)-1b | 99 | 99 ee |
| 16 | 3b |
|
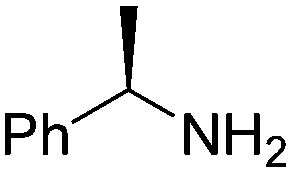
(SP)-19 | 72 | 96 de |

To further explore the scope of this transformation we submitted diastereomerically pure aminophosphines 5a and 5b (the synthetic precursors of aminophosphines 1a and 1b) to acidic hydrolysis in MeOH/H2O mixtures (Scheme 2). The reaction was slower than that achieved using the parent compounds 1a and 1b;13 however, again, an increase in the reaction temperature and a longer reaction time produced the enantiomerically pure phosphinous acids 3a and 3b in 84% and 66% yields respectively. The hydrolysis of 5a and 5b is a practical approach to prepare the corresponding phosphinous acids; furthermore, it allows the recovery of the chiral auxiliary.
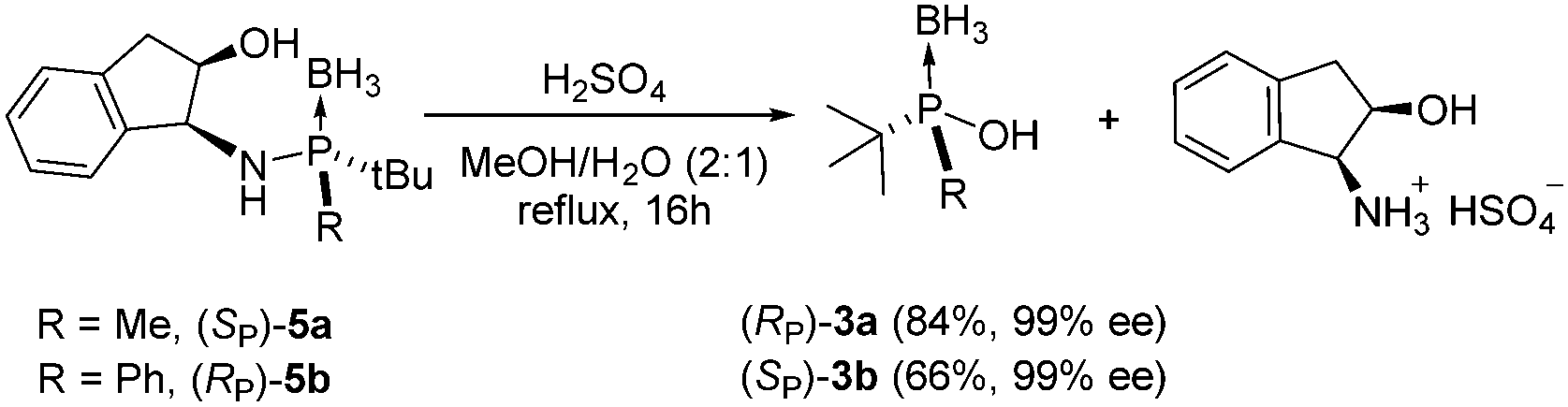 | ||
| Scheme 2 Acidolysis of N-secondary aminophosphines and recovery of the chiral auxiliary. | ||
Optically pure P-stereogenic phosphinous acid boranes are attractive synthetic intermediates; however, they have barely been used in ligand synthesis.14 While optically enriched 3b has been prepared independently by Pietrusiewicz and Buono via resolution or H-menthylphosphinate technology, this is the first time that phosphinous acid 3a has been reported. It is known that mesyl-activated phosphinous acids undergo effective nucleophilic reductions in the presence of NaBH4 with inversion at the phosphorus center,14 and we speculated whether this process could be extended to nucleophiles larger than hydride. Hence, phosphinous acid (SP)-3a was treated with Ms2O in the presence of triethylamine, and the reactions of the resulting mixed anhydride with several nucleophiles were studied (Table 2).15
Initial experiments using ammonia as the nucleophile indicated that, in solution, the phosphinyl-mesyl anhydride underwent slow racemization. Fortunately, a judicious choice of solvent and lowering of the reaction temperature to −20 °C permitted the nucleophilic substitution with ammonia in an almost completely stereospecific fashion. This produced (RP)-1a in 99% yield and 96% ee with the inverted configuration at the P-center (Table 2, entry 1). Importantly, primary amines also acted as efficient nucleophiles in this process, producing the corresponding aminophosphines in satisfactory yield and excellent enantiomeric excess (Table 2, entries 2–5). Chiral primary α-branched amines like (S) and (R)-1-phenylethylamine and phenylglycinamide also yielded the substitution products (RP)-10, (RP)-11 and (RP)-12 in 95–98% diastereomeric excess (Table 2, entries 6–8). A cyclic secondary amine like pyrrolidine and an aromatic amine like p-anisidine also efficiently produced the substitution products in 98 and 91% ee (Table 2, entries 9 and 10). In contrast, dibenzylamine was not good enough as a nucleophile and did not afford the expected aminophosphine (Table 2, entry 11). Also, oxygen nucleophiles failed to provide substitution products (Table 2, entry 12). In contrast, benzylthiol and thiophenol did provide the corresponding sulfides 17 and 18, albeit in low yield (22 and 36% respectively) (Table 2, entries 13 and 14). We attributed these low yields of isolation to the instability of these molecules. Finally, we tested the tert-butylphenylphosphinous acid borane 3b as the electrophile. Reaction with ammonia and (R)-phenylethylamine provided the corresponding aminophosphines 1b and 19 with excellent optical and diastereomeric purity (Table 2, entries 15 and 16).
The hydrolysis of aminophosphine boranes combined with the substitution on the resulting phosphinous acid represents a novel means of obtaining P*-ligands. To highlight the beneficial impact that this methodology could have in asymmetric catalysis, we followed the procedure shown in Table 2 to prepare a novel P-stereogenic N-phosphinooxazoline ligand and its corresponding cationic iridium complex (Scheme 3). Activation of (SP)-3a with mesyl anhydride and triethylamine and reaction with aminooxazoline 20 provided the borane-protected ligand 21 in 43% yield and in >98% dr, as determined by 1H NMR. Borane was removed in neat pyrrolidine at 90 °C.16 We found that the approach of using neat pyrrolidine was superior to that of existing borane-deprotection methods like DABCO or neat diethylamine. Thus, from 21 and using a one pot-three reaction sequence, the cationic iridium complex 22 was obtained as an orange solid in 89% yield.
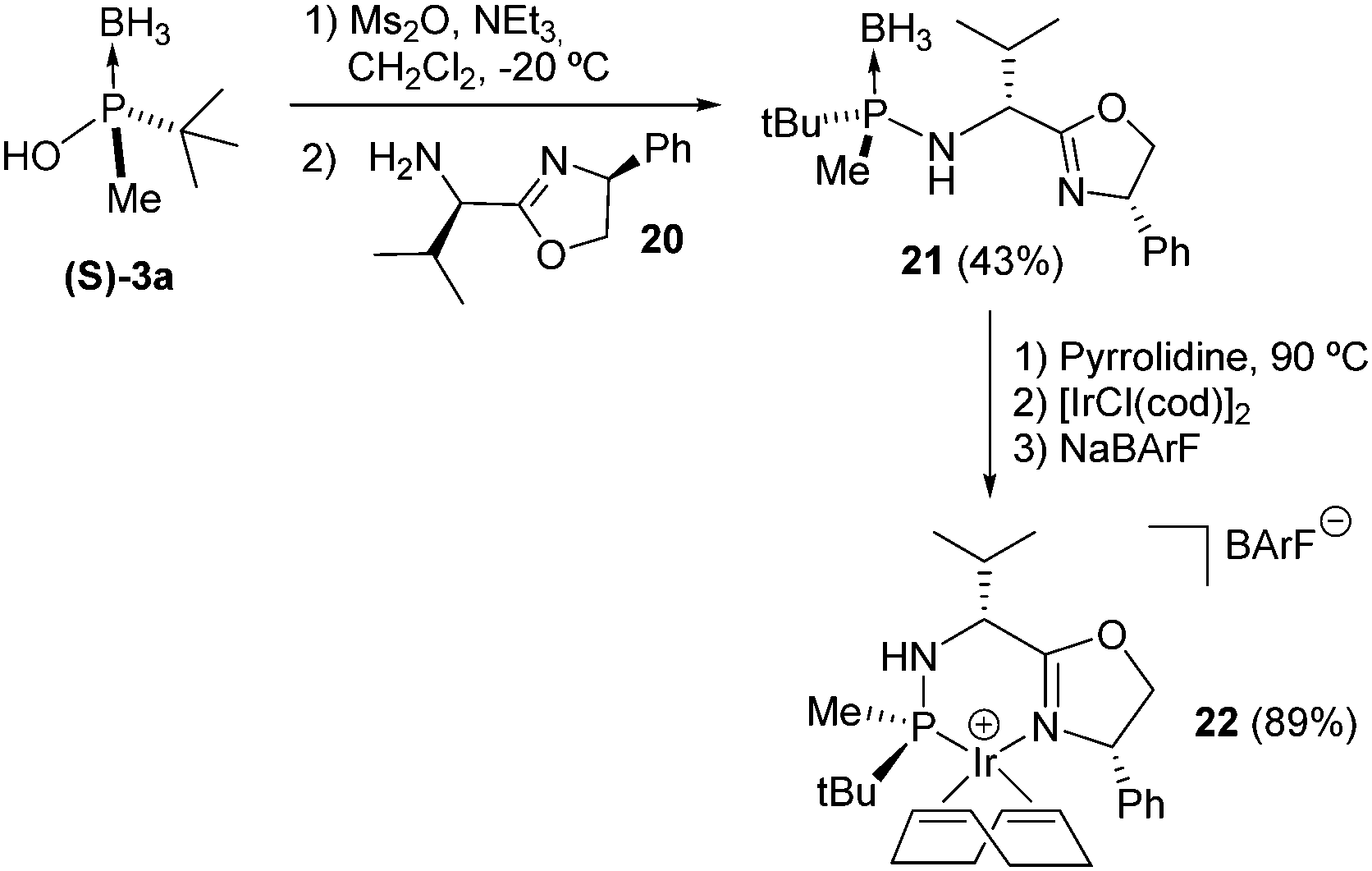 | ||
| Scheme 3 Synthesis of the P-stereogenic N-phosphino-oxazoline iridium complex. | ||
With the iridium complex 22 in hand, we tested its performance in the asymmetric hydrogenation of α,β-unsaturated esters (Table 3).17,18 Reduction of ethyl trans-β-methylcinnamate under standard non-optimized conditions (50 bar of hydrogen in dichloromethane with 1 mol% of 22 as the catalyst) produced the (R) hydrogenated product in 95% ee (Table 3, entry 1). Under the same reaction conditions, hydrogenation of the isopropyl and cyclohexyl β-substituted cinnamates afforded the reduced products in 97% ee (Table 3, entries 2 and 3). Finally, para-methyl-substituted methylcinnamate afforded the reduced compound with complete selectivity (>99% ee, Table 3, entry 4).

In summary, we have shown that the acid hydrolysis of bulky primary and secondary aminophosphine boranes occurs in a completely stereospecific manner with inversion of the configuration at the P-center to yield the corresponding optically pure phosphinous acid boranes. Also, we have demonstrated that, upon activation, phosphinous acid boranes undergo stereospecific nucleophilic substitution reactions at the P-center with amine nucleophiles. The potential of this process has been demonstrated with the synthesis of a P-stereogenic phosphinooxazoline ligand which has been applied to the asymmetric Ir-catalyzed hydrogenation of trans-β-alkylcinnamates, achieving selectivities of up to 99% ee.
We thank financial support from the Spanish Ministerio de Economia y Competividad (CTQ2014-56361-P and CTQ2013-40568P), the IRB Barcelona, the Generalitat de Catalunya (2014SGR670), the ICREA Foundation (M. Diéguez and O. Pàmies ICREA Academia awards). S.O. thanks the Generalitat de Catalunya for a FI fellowship. A. F.-G. thanks the CONACYT for a postdoctoral fellowship.
Notes and references
- Phosphorous Ligands in Asymmetric Catalysis, ed. A. Börner, Wiley-WCH, Weinheim, 2008, vol. I–III Search PubMed.
- For general reviews on P-stereogenic phosphine-ligands, see: (a) A. Grabulosa, P-Stereogenic Ligands in Asymmetric Catalysis, RSC Publishing, Cambridge, 2011 Search PubMed; (b) A. Grabulosa, J. Granell and G. Muller, Coord. Chem. Rev., 2007, 251, 25 CrossRef CAS; (c) O. I. Kolodiazhnyi, Top. Curr. Chem., 2015, 360, 161 CrossRef CAS PubMed . For recent examples on the synthesis of P-stereogenic phosphines, see: ; (d) D. Gatineau, D. H. Nguyen, D. Hérault, N. Vanthuyne, J. Leclaire, L. Giordano and G. Buono, J. Org. Chem., 2015, 80, 4132 CrossRef CAS PubMed; (e) Z. S. Han, N. Goyal, M. A. Herbage, J. D. Sieber, B. Qu, Y. Xu, Z. Li, J. T. Reeves, J. N. Desrosiers, S. Ma, N. Grinberg, H. Lee, H. P. R. Mangunuru, Y. Zhang, D. Krishnamurthy, B. Z. Lu, J. J. Song, G. Wang and C. H. Senanayake, J. Am. Chem. Soc., 2013, 135, 2474 CrossRef CAS PubMed; (f) K. Nikitin, K. V. Rajendran, H. Müller-Bunz and D. G. Gilheany, Angew. Chem., Int. Ed., 2014, 53, 1906 CrossRef CAS PubMed; (g) R. den Heeten, B. H. G. Swennenhuis, P. W. N. M. van Leeuwen, J. G. de Vries and P. C. Kamer, Angew. Chem., Int. Ed., 2008, 47, 6602 CrossRef CAS PubMed; (h) Q. Xu, C. Q. Zhao and L. B. Han, J. Am. Chem. Soc., 2008, 130, 12648 CrossRef CAS PubMed.
- For recent examples of the use of bulky P-stereogenic phosphines in asymmetric catalysis, see: (a) T. Imamoto, K. Tamura, Z. Zhang, Y. Horiuchi, M. Sugiya, K. Yoshida, A. Yanagisawa and I. D. Gridnev, J. Am. Chem. Soc., 2012, 134, 1754 CrossRef CAS PubMed; (b) W. Tang, B. Qu, A. G. Capacci, S. Rodriguez, X. Wei, N. Haddad, B. Narayanan, S. Ma, N. Grinberg, N. K. Yee, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2010, 12, 176 CrossRef CAS PubMed; (c) H. Landert, F. Spindler, A. Wyss, H. U. Blaser, B. Pugin, Y. Ribourduoille, B. Gschwend, B. Ramalingam and A. Pfaltz, Angew. Chem., Int. Ed., 2010, 49, 6873 CrossRef CAS PubMed; (d) I. H. Chen, L. Yin, W. Itano, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 11664 CrossRef CAS PubMed; (e) H. Ito, T. Okura, K. Matsuura and M. Sawamura, Angew. Chem., Int. Ed., 2010, 49, 560 CrossRef CAS PubMed; (f) X. Wang and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 19080 CrossRef CAS PubMed; (g) G. Xu, W. Fu, G. Liu, C. H. Senanayake and W. Tang, J. Am. Chem. Soc., 2014, 136, 570 CrossRef CAS PubMed; (h) G. Liu, X. Liu, Z. Cai, G. Jiao, G. Xu and W. Tang, Angew. Chem., Int. Ed., 2013, 52, 4235 CrossRef CAS PubMed.
- (a) A. R. Muci, K. R. Campos and D. A. Evans, J. Am. Chem. Soc., 1995, 117, 9075 CrossRef CAS; (b) T. Imamoto, J. Watanabe, Y. Wada, H. Masuda, H. Yamada, H. Tsuruta, S. Matsukawa and K. Yamaguchi, J. Am. Chem. Soc., 1998, 120, 1635 CrossRef CAS; (c) W. Tang, W. Wang and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 943 CrossRef CAS PubMed; (d) J. J. Gammon, V. H. Gessner, G. R. Barker, J. Granander, A. C. Whitwood, C. Strohmann, P. O’Brien and B. Kelly, J. Am. Chem. Soc., 2010, 132, 13922 CrossRef CAS PubMed; (e) W. Tang and X. Zhang, Angew. Chem., Int. Ed., 2002, 41, 1612 CrossRef CAS.
- (a) T. Imamoto, K. Sugita and K. Yoshida, J. Am. Chem. Soc., 2005, 127, 11934 CrossRef CAS PubMed; (b) K. Tamura, M. Sugiya, K. Yoshida, A. Yanagisawa and T. Imamoto, Org. Lett., 2010, 12, 4400 CrossRef CAS PubMed.
- (a) J. M. Brown and J. C. P. Laing, J. Organomet. Chem., 1997, 529, 435 CrossRef CAS; (b) F. Maienza, F. Spindler, M. Thommen, B. Pugin, C. Malan and A. Mezzetti, J. Org. Chem., 2002, 67, 5239 CrossRef CAS PubMed; (c) E. V Jennings, K. Nikitin, Y. Ortin and D. G. Gilheany, J. Am. Chem. Soc., 2014, 136, 16217 CrossRef PubMed.
- Buono and co-workers reported the stereodivergent and stereoselective hydrolysis of an unprotected aminophosphine to yield the corresponding secondary phosphine oxide with up to 91% ee, see: A. Leyris, D. Nuel, L. Giordano, M. Achard and G. Buono, Tetrahedron Lett., 2005, 46, 8677 CrossRef CAS.
- T. Imamoto, Y. Saitoh, A. Koide, T. Ogura and K. Yoshida, Angew. Chem., Int. Ed., 2007, 46, 8636 CrossRef CAS PubMed.
- (a) S. Jugé, M. Stephan, J. A. Laffitte and J. P. Genet, Tetrahedron Lett., 1990, 31, 6357 CrossRef; (b) C. Darcel, J. Uziel and S. Jugé, in Phosphorus Ligands in Asymmetric Catalysis, ed. A. Börner, Wiley-WCH, Weinheim, 2008, vol. III, pp. 1211–1233 Search PubMed.
- C. Bauduin, D. Moulin, E. B. Kaloun, C. Darcel and S. Jugé, J. Org. Chem., 2003, 68, 4293 CrossRef CAS PubMed.
- (a) E. Cristóbal-Lecina, P. Etayo, S. Doran, M. Revés, P. Martín-Gago, A. Grabulosa, A. R. Costantino, A. Vidal-Ferran, A. Riera and X. Verdaguer, Adv. Synth. Catal., 2014, 356, 795 CrossRef; (b) T. León, M. Parera, A. Roglans, A. Riera and X. Verdaguer, Angew. Chem., Int. Ed., 2012, 51, 6951 CrossRef PubMed.
- CCDC 1412790.
- (a) H. Zijlstra, T. León, A. de Cózar, C. Fonseca Guerra, D. Byrom, A. Riera, X. Verdaguer and F. M. Bickelhaupt, J. Am. Chem. Soc., 2013, 135, 4483 CrossRef CAS PubMed; (b) T. León, A. Riera and X. Verdaguer, J. Am. Chem. Soc., 2011, 133, 5740 CrossRef PubMed.
- (a) M. Stankevic and K. M. Pietrusiewicz, J. Org. Chem., 2007, 72, 816 CrossRef CAS PubMed; (b) D. Moraleda, D. Gatineau, D. Martin, L. Giordano and G. Buono, Chem. Commun., 2008, 3031 RSC; (c) D. Gatineau, L. Giordano and G. Buono, J. Am. Chem. Soc., 2011, 133, 10728 CrossRef CAS PubMed.
- From this point forward the opposite enantiomer of the phosphinous acid borane (SP)-5a was used.
- G. C. Lloyd-Jones and N. P. Taylor, Chem. – Eur. J., 2015, 21, 5423 CrossRef CAS PubMed.
- For reviews, see: (a) S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402 CrossRef CAS PubMed; (b) T. L. Church and P. G. Andersson, Coord. Chem. Rev., 2008, 252, 513 CrossRef CAS; (c) D. H. Woodmansee and A. Pfaltz, Chem. Commun., 2011, 47, 7912 RSC; (d) Y. Zhu and K. Burgess, Acc. Chem. Res., 2012, 45, 1623 CrossRef CAS PubMed; (e) J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130 CrossRef CAS PubMed.
- For recent successful examples of Ir-hydrogenation of this substrate class, see: (a) J. Q. Li, X. Quan and P. G. Andersson, Chem. – Eur. J., 2012, 18, 10609 CrossRef CAS PubMed; (b) D. H. Woodmansee, M. A. Müller, L. Tröndlin, E. Hörmann and A. Pfaltz, Chem. – Eur. J., 2012, 18, 13780 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Stephen L. Buchwald on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, characterization data and NMR spectra for new compounds. CCDC 1412790. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc07504a |
| § Present address: Facultad de Ciencias, Universidad Antonio Nariño, Bogotá, Colombia. |
| This journal is © The Royal Society of Chemistry 2015 |