
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled generation of highly saddled (porphyrinato)iron(III) iodide, tri-iodide and one-electron oxidized complexes†
Dipankar
Sahoo
and
Sankar Prasad
Rath
*
Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur-208016, India. E-mail: sprath@iitk.ac.in; Fax: +91 512 259 7436; Tel: +91 512 259 7251
First published on 21st September 2015
Abstract
For the first time, three iron(III) porphyrinato complexes have been synthesized selectively by varying the iodine concentration in the reaction mixture which eventually forms both five and six coordinate complexes with iodide and/or tri-iodide as axial ligands. Combined analysis using single crystal X-ray structure determination, and Mössbauer, 1H NMR and EPR studies as well as VT magnetic studies has revealed the admixed-intermediate (iodo complex), pure intermediate (tri-iodide complex) and high-spin (1e-oxidized complex) states of iron.
Hemoproteins serve many diverse biological functions through the nearly identical heme prosthetic group as a consequence of the subtle coordination and redox chemistry apparent for iron porphyrins.1–4 A variety of geometries, and oxidation and spin states of iron porphyrins, which are critical intermediates in the catalytic cycles of both biological and abiological systems, have been reproduced systematically using chemical models.1–11 The spin states of five-coordinate iron(III) porphyrins are controlled mainly by the ligand field strength of axial ligands.1,4–7 While most of the anionic ligands such as halides and hydroxides lead to the formation of high-spin (S = 5/2) complexes, extremely weak field ligands such as ClO4 and SbF6 give the complexes with quantum mechanical spin admixed states with varying proportions of pure S = 3/2 and S = 5/2 states. In fact, the degree of the S = 3/2 contribution fairly correlates with the ligand field strength of the anionic axial ligands when other factors are identical. In addition, the deformation of the porphyrin ring has also been known to influence the electronic structures of the iron(III) porphyrin complexes.4–7 We report here the synthesis, X-ray structure and properties of three saddle distorted Fe(III) porphyrins generated via the controlled addition of iodine in the reaction mixture.
Free base octaethyltetraarylporphyrin 1 (Scheme 1) was prepared according to the literature procedure,12 and the metal was inserted using excess FeI2 in N,N-dimethylformamide under nitrogen. While washing the chloroform solution of the metal inserted complex with 1% HI in air forms the iodide complex 2, the addition of 5% HI solution along with one equivalent iodine resulted in the formation of the tri-iodide complex 3. Moreover, the addition of excess solid I2 (5 equiv.) to either iodide (2) or tri-iodide complex (3) has produced the identical one-electron oxidized six-coordinate complex 4 where both iodide and tri-iodide occupy the fifth and sixth positions. Thus, 2, 3 and 4 have been prepared by selectively varying the concentration of I2 in the reaction mixture (Scheme 1). All the molecules have been isolated as a crystalline solid in high yields and two of them (2 and 3) are also structurally characterized. The UV-visible spectrum of 2 shows the Soret band at 416 nm in CHCl3, while in 3, the band shifted via a small blue shift to 413 nm along with a weak shoulder at 440 nm (Fig. 1). However, for the 1e-oxidized complex 4, a blue-shifted broad Soret band appeared at 408 nm along with an intense shoulder peak at 442 nm. Typically, the electronic spectrum characteristics of a radical cation species are a new band at low energy and a dramatically broadened, blue-shifted Soret band relative to the unoxidized species.8 Here, 4 displays the characteristic features of π-cation radicals. However, the near-IR band, which is associated with the formation of a dimeric π-cation radical, has not been observed in 4 due to the formation of the six-coordinate complex. The oxidized complex showed the IR marker bands8 characteristic for a porphyrin π-cation radical at 1596 and 1448 cm−1 which indicate the formation of a π-cation radical in the complex (Fig. S1, ESI†). The electrospray ionization mass spectrometry (ESI-MS) showed intense peaks at m/z 1131.4481 for [2]+, 1385.6151 for [3]+, and 1512.1478 for [4]+ (Fig. S2–S4, ESI†) confirming the formation of the complexes. The isotopic distribution patterns of the experimental mass were also nicely correlated with the calculated patterns.
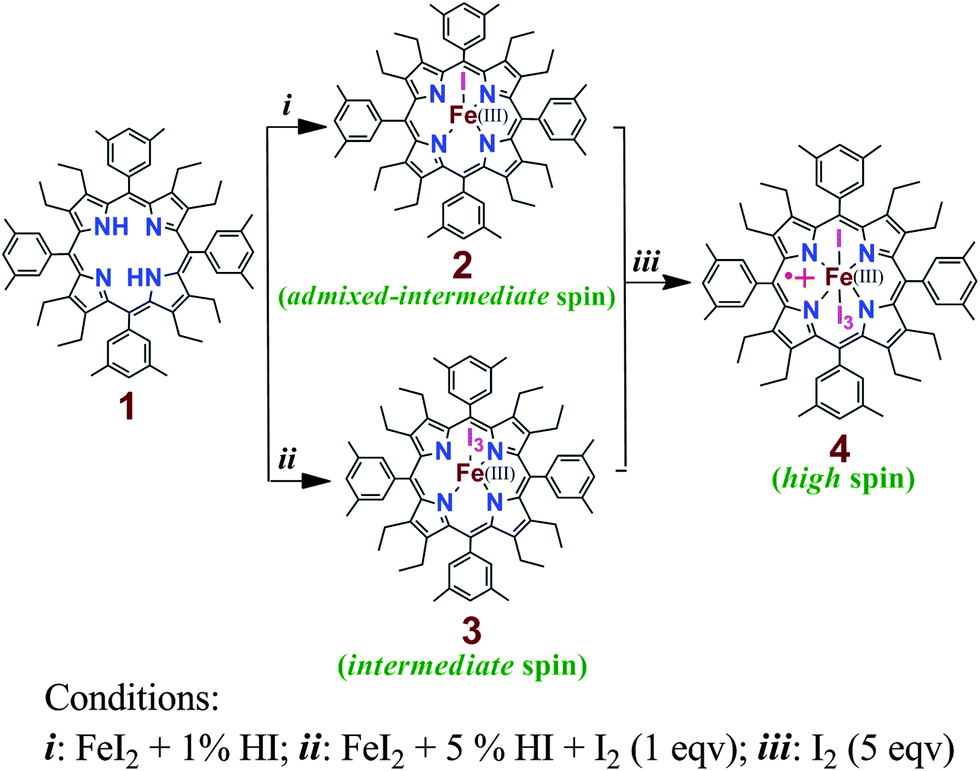 | ||
| Scheme 1 | ||
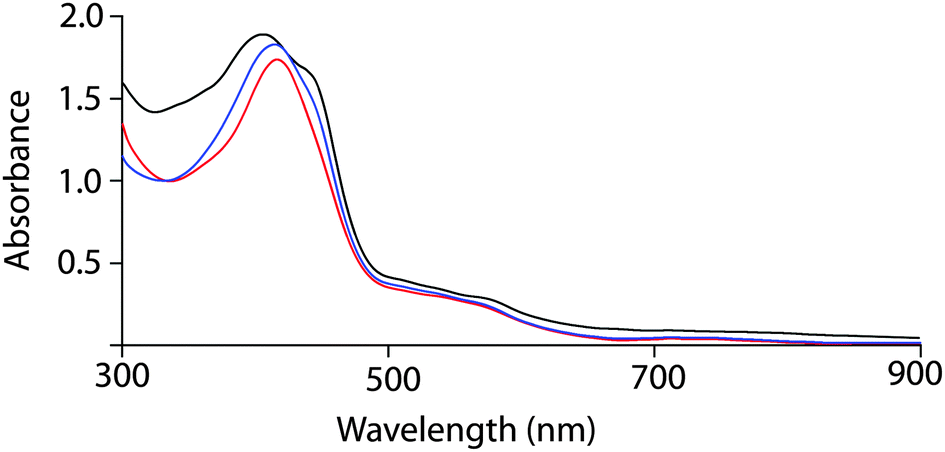 | ||
| Fig. 1 UV-visible spectra (in CHCl3 at 295 K) using polycrystalline samples of 2 (red line), 3 (blue line) and 4 (black line). | ||
Dark-purple crystals of 2 and 3 were grown by slow diffusion of cyclohexane and acetonitrile, respectively, into the solution of the respective complex in chloroform at room temperature in air.13 Dark-purple crystals of the oxidized complex 4 were also obtained by slow diffusion of hexane into the chloroform solution of the complex. However, the highly disordered nature of the I3 axial ligand failed to provide a good quality structure of the complex. The perspective views of 2 and 3 are shown in Fig. 2 (and see Fig. S5 and S6 (ESI†) for the molecular packing diagrams). In the X-ray structure (Tables S1 and S2, ESI†), the iron centers have a five-coordinate square-pyramidal geometry and have shown considerable doming of the porphyrin cores. The Fe–I bond undergoes an extensive elongation from a distance of 2.5236(10) Å in 2 to 2.7503(19) Å in the tri-iodide complex 3 which is also indicative of a weaker ligand field strength of the tri-iodide ion. Also, the average Fe–Np distances in the two types of complexes are different: 1.988(4) Å in 2 and 1.947(8) Å in 3. As can be seen, the Fe–I distance increases as the bond distance of Fe–Np decreases. The displacement of the iron center from the mean plane of the C20N4 porphyrinato core (ΔFe24) is contracted from 0.36 Å (in 2) to 0.21 Å (in 3). All these structural features are characteristic for having admixed-intermediate and pure intermediate spin (S = 3/2) states of iron, respectively, in 2 and 3.4–11 In contrast, iron centers are mostly high-spin in all the previously reported iodo complexes (Table S3, ESI†) that are structurally characterized.9 Also, porphyrin rings are highly distorted in 2 and 3 and assume a nearly pure saddle shape (Fig. S7 and S8, ESI†).
 | ||
| Fig. 2 Perspective views (at 100 K) of (A) 2 and (B) 3 showing 50% thermal contours for all non-hydrogen atoms (H atoms have been omitted for clarity). Selected bond distances (Å) and angles (deg) for 2: Fe1–N1, 1.977(4); Fe1–N2, 1.999(4); Fe1–N3, 1.976(4); Fe1–N4, 2.001(4); Fe1–I1, 2.5236(10); N1–Fe1–N2, 88.08(16); N1–Fe1–N4, 88.36(16); N2–Fe1–N4, 151.14(17); N3–Fe1–N1, 167.06(17); N3–Fe1–N2, 88.36(17); N3–Fe1–N4, 88.76(17); N1–Fe1–I1, 96.26(12); N2–Fe1–I1, 105.51(12); N3–Fe1–I1, 96.69(12); N4–Fe1–I1, 103.35(12). For 3: Fe1–N1, 1.932(6); Fe1–N2, 1.975(10); Fe1–N3, 1.951(9); Fe1–I1, 2.7503(19); I1–I2, 3.0164(14); N1–Fe1–N2, 89.53(19); N3–Fe1–N1, 89.42(19); N3–Fe1–N2, 158.3(4); N1–Fe1–I1, 92.8(2); N2–Fe1–I1, 101.4(3); N3–Fe1–I1, 100.4(3); I1–I2–I3, 173.87(5). | ||
Mössbauer parameters are one of the most powerful probes to determine the spin state of the iron(III) porphyrins.5,7a,8Fig. 3 compares the Mössbauer spectra of the microcrystalline samples of 2, 3, and 4 at 100 K. Complexes 2 and 3 have displayed a doublet with a large quadrupole-splitting [δ(ΔEq): 0.35 (3.42) mm s−1 for 2 and 0.37 (3.80) mm s−1 for 3] characteristic of an admixed-intermediate and a pure intermediate (S = 3/2) state of iron, respectively. In contrast, 1e-oxidized complex 4 exhibits a broad doublet with a small quadrupole-splitting from which the IS and QS values are determined to be 0.27 and 0.74 mm s−1, respectively, which fall within the range of the parameters known for high-spin (S = 5/2) Fe(III) porphyrins.
 | ||
| Fig. 3 Mössbauer spectra of (A) 2, (B) 3, and (C) 4 at 100 K. | ||
To obtain much conclusive evidence for the spin state, the EPR spectroscopic measurements were carried out for the complexes in both solid and solution phases (Fig. S9, ESI†). The spectra were carefully simulated (a representative simulated spectrum is shown in Fig. S10, ESI†). All the spectra are axially symmetric with g⊥ = 4.24 and g∥ = 1.99 for 2, and g⊥ = 4.05 and g∥ = 2.01 for 3 at 77 K in the solid state. Similar g values are also obtained for the molecules in solution. In the case of 2, hyperfine coupling of 50.8 G has clearly been observed at g = 2 with the axially coordinated iodide. The contributions of the S = 3/2 spin state can be estimated as (6 − g⊥)/2,5 which are calculated to be 88% and 98% for 2 and 3, respectively. Consistent with the X-ray structure, and SQUID and Mössbauer results, 3 shows a quite pure S = 3/2 spin state.
1H NMR spectroscopy is a valuable tool to distinguish different spin states of iron(III) porphyrins in solution.4,7,10,11,14Fig. 4 shows the spectra of 2, 3 and 4 recorded at 295 K in CDCl3. The 1H NMR chemical shifts between 2 and 3 exhibit some differences depending on the axial ligand strengths. For complex 2, four methylene proton signals are observed at 48.81, 32.15, 30.15, and 15.06 ppm (average 31.54 ppm) which are similar to those of the five-coordinate iron(III) iodo complex with the admixed-intermediate spin states reported earlier.14a In complex 3, however, four methylene peaks are observed at 42.25, 14.09, 12.85, and 9.45 ppm (average 19.66 ppm) and are characteristic of pure intermediate spin states.7,11 Thus, upon moving from an iodo to a tri-iodide complex, the spin state of iron has been changed from a admixed-intermediate state to a pure intermediate state.
 | ||
| Fig. 4 1H NMR spectra of (A) 2, (B) 3, and (C) 4 at 295 K in CDCl3. | ||
For a one-electron oxidized complex 4, four methylene proton signals are observed at 87.06, 14.07 and 10.28 (2) ppm which are, however, characteristic of the high-spin state of iron.4,7a Interestingly, the ortho and para proton signals of the phenyl substituents are observed at 30.29, 28.09 and 36.65 ppm, respectively, (Fig. 4C) which indicates the presence of a2u porphyrin π-cation radicals.14b A characteristic feature of this type of radical is the large spin densities on the meso carbons which induce large isotropic shifts of the meso phenyl protons in the 1H NMR spectrum as exemplified by the extremely downfield-shifted ortho and para protons and upfield-shifted meta signals.4,14 The result also indicates that 4 has a negative spin density at the meso carbon atoms caused by the antiferromagnetic coupling between the paramagnetic iron and the a2u radical spin.14 The oxidation has also induced a wider separation between up-field- and down-field-shifted CH2 signals which can be explained to be due to the formation of π-cation radicals along with a change of iron spin state from S = 3/2 to 5/2.
Variable-temperature magnetic susceptibility measurements has been carried out in the solid state for 1e-oxidized complex 4, and was fit (Fig. S11, ESI†) using the software PHI.15 The iron(III) center was treated as a high spin (S = 5/2) with a g value set at 2.0 and the presence of a small amount of residual iron(III) impurity was also taken into consideration. The coupling between the iron spin and the porphyrin radical does show a significant value (−JFe–r = 62.8 cm−1). The inter-ring coupling that is closely related only to the degree of the ring overlap8a has not been considered due to the six-coordinate nature of the complex. Also, the near-IR band, which is associated with the formation of a dimeric π-cation radical,8 has not been observed in 4. The a2u radical wave function has a large amplitude at the pyrrole nitrogens, and thus it is expected that the a2u orbital overlaps strongly with the iron d-orbitals which resulted in a larger iron–radical coupling.14,16
To gain insight into the origin of the spin-state, we ran a series of density functional theory calculations on 3 and 4 (see the ESI† for details) considering both high (S = 5/2) and intermediate spin (S = 3/2) states of iron in each case. Geometry optimisations were carried out using respective spin multiplicities: quartet and sextet states for 3 while triplet (considering an antiferromagnetic interaction between the π-cation radical and the intermediate spin of iron(III)) and quintet (considering an antiferromagnetic coupling between the π-cation radical and the high-spin of iron(III)) states for 4. The optimized geometries of 3 and 4 are shown in Fig. 5. As can be seen, the optimized geometries of 43 match the crystal structure coordinates reasonably well. Also, 3 has been found to be stabilized in an intermediate-spin (IS) state with ΔGHS/IS = 10.63 kcal mol−1 as compared to the high-spin (HS) state. In contrast, the addition of two axial ligands iodide and tri-iodide changes the spin state ordering in 4 and stabilizes the high-spin state (ΔGHS/IS = −6.7 kcal mol−1) of iron.
 | ||
| Fig. 5 UB3LYP/BS1 optimized geometries of 4,63 and 3,54 with bond lengths (r) in Å. ΔFe24 is the average displacement (in Å) of the metal from the least-square plane of the C20N4 porphyrinato core. Free energies ΔGHS/IS are relative to the intermediate spin state in kcal mol−1. | ||
Studies on model hemes suggest that the intermediate-spin state is critically dependent on the axial ligand strength. Tri-iodide is a weaker axial ligand compared to iodide, which is, however, responsible for the stabilization of the admixed intermediate and pure intermediate spin states of iron in 2 and 3, respectively, as demonstrated here. Both the molecules, although looking very similar, are carefully isolated and structurally characterized which enable us to investigate their spectroscopic identities that were missing so far. Moreover, the one-electron oxidized six-coordinate complex 4 stabilizes the high-spin state of iron with iodide and tri-iodide as axial ligands and has been formulated as the iron(III) porphyrin π-cation radical. A variable temperature magnetic study in solids has shown a strong antiferromagnetic coupling between iron and radical spins in the oxidized complex 4. Furthermore, computational calculations clearly support the experimentally assigned spin state.
For the first time, three iron(III) porphyrinato complexes have been synthesized selectively just by slight variations of the iodine concentration in the reaction mixture which eventually forms both five- and six-coordinate complexes with iodide and/or tri-iodide as axial ligands. Single crystal X-ray structure determination, and Mössbauer, 1H NMR and EPR studies as well as VT magnetic studies have revealed their electronic structure and properties. Iodine is not known to be a strong oxidant; however, axial coordination of iodide/tri-iodide in 2/3 enables the facial oxidation of the complexes using iodine to form the iron(III) porphyrin π-cation radical 4. The present work also highlights the various roles of iodine and opportunities therein in chemical synthesis. Further work is in progress.
We thank the Science and Engineering Research Board (SERB), India, and CSIR, New Delhi, for financial support. We also thank Dr Matthew G. Quesne for a useful discussion on DFT.
Notes and references
- W. R. Scheidt, in The Porphyrin Handbook, ed. K. M. Kadishi, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 3, pp. 49–112 Search PubMed.
- (a) J. A. Shelnutt, X.-Z. Song, J.-G. Ma, S.-L. Jia, W. Jentzen and C. J. Medforth, Chem. Soc. Rev., 1998, 27, 31–42 RSC; (b) M. O. Senge, Chem. Commun., 2006, 243 RSC.
- (a) F. A. Walker, Chem. Rev., 2004, 104, 589–615 CrossRef CAS PubMed; (b) F. A. Walker, Coord. Chem. Rev., 1999, 185–186, 471–534 CrossRef CAS.
- (a) A. Ikezaki, Y. Ohgo and M. Nakamura, Coord. Chem. Rev., 2009, 253, 2056–2069 CrossRef CAS; (b) M. Nakamura, Coord. Chem. Rev., 2006, 250, 2271–2294 CrossRef CAS.
- (a) R. Weiss, A. Gold and J. Terner, Chem. Rev., 2006, 106, 2550–2579 CrossRef CAS PubMed; (b) Y. Ling and Y. Zhang, J. Am. Chem. Soc., 2009, 131, 6386–6388 CrossRef CAS PubMed.
- (a) C. A. Read and F. Guiset, J. Am. Chem. Soc., 1996, 118, 3281–3282 CrossRef; (b) R. J. Cheng, P. Y. Chen, P. R. Gau, C. C. Chen and S. M. Peng, J. Am. Chem. Soc., 1997, 119, 2563–2569 CrossRef CAS.
- (a) D. Sahoo, M. G. Quesne, S. P. de Visser and S. P. Rath, Angew. Chem., Int. Ed., 2015, 54, 4796–4800 CrossRef CAS PubMed; (b) S. Bhowmik, S. K. Ghosh and S. P. Rath, Chem. Commun., 2011, 47, 4790–4792 RSC; (c) S. K. Ghosh, R. Patra and S. P. Rath, Inorg. Chem., 2010, 49, 3449–3460 CrossRef CAS PubMed.
- (a) M. Li, T. J. Neal, G. R. A. Wyllie, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 2010, 49, 8078–8085 CrossRef CAS PubMed; (b) M. Li, T. J. Neal, G. R. A. Wyllie, A. G. Oliver, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 2011, 50, 9114–9121 CrossRef CAS PubMed; (c) S. Hu and T. G. Spiro, J. Am. Chem. Soc., 1993, 115, 12029–12034 CrossRef CAS.
- (a) Y. Ohgo, S. Neya, M. Takahashi, M. Takeda, N. Funasaki and M. Nakamura, Chem. Lett., 2003, 32, 526–527 CrossRef CAS; (b) K. Hatano and W. R. Scheidt, Inorg. Chem., 1979, 18, 877–879 CrossRef CAS; (c) Y. Ohgo, S. Neya, T. Ikeue, M. Takahashi, M. Takeda, N. Funasaki and M. Nakamura, Inorg. Chem., 2002, 41, 4627–4629 CrossRef CAS PubMed.
- (a) R. Patra, D. Sahoo, S. Dey, D. Sil and S. P. Rath, Inorg. Chem., 2012, 51, 11294–11305 CrossRef CAS PubMed; (b) R. Patra, S. Bhowmik, S. K. Ghosh and S. P. Rath, Dalton Trans., 2010, 39, 5795–5806 RSC; (c) R. Patra, A. Chaudhury, S. K. Ghosh and S. P. Rath, Inorg. Chem., 2008, 47, 8324–8335 CrossRef CAS PubMed; (d) R. Patra and S. P. Rath, Inorg. Chem. Commun., 2009, 515–519 CrossRef CAS.
- (a) D. Sil and S. P. Rath, Dalton Trans., 2015, 44, 16195–16211 RSC; (b) M. A. Sainna, D. Sil, D. Sahoo, B. Martin, S. P. Rath, P. Comba and S. P. de Visser, Inorg. Chem., 2015, 54, 1919–1930 CrossRef CAS PubMed; (c) D. Sil, F. S. T. Khan and S. P. Rath, Inorg. Chem., 2014, 53, 11925–11936 CrossRef CAS PubMed; (d) S. K. Ghosh, S. Bhowmik, D. Sil and S. P. Rath, Chem. – Eur. J., 2013, 19, 17846–17859 CrossRef CAS PubMed; (e) S. Bhowmik, S. Dey, D. Sahoo and S. P. Rath, Chem. – Eur. J., 2013, 19, 13732–13744 CrossRef CAS PubMed; (f) S. Bhowmik, S. K. Ghosh, S. Layek, H. C Verma and S. P. Rath, Chem. – Eur. J., 2012, 18, 13025–13037 CrossRef CAS PubMed; (g) S. K. Ghosh and S. P. Rath, J. Am. Chem. Soc., 2010, 132, 17983–17985 CrossRef CAS PubMed; (h) S. K. Ghosh, R. Patra and S. P. Rath, Inorg. Chim. Acta, 2010, 363, 2791–2799 CrossRef CAS.
- K. M. Barkigia, M. D. Berber, J. Fajer, C. J. Medforth, M. W. Renner and K. M. Smith, J. Am. Chem. Soc., 1990, 112, 8851–8857 CrossRef CAS.
- Crystal data for 2: orthorhombic, space group Pbca, Z = 8, a = 19.736(5) Å, b = 24.456(5) Å, c = 31.304(5) Å, V = 15109(5) Å3. dcalcd = 1.014 Mg m−3. R1 = 0.0888, wR2 (all data) = 0.2583. Goodness of fit on F2 = 1.034. For 3 orthorhombic, space group Pnma, Z = 1, a = 23.387(5) Å, b = 17.301(4) Å, c = 18.913(4) Å, V = 7653(3) Å3. dcalcd = 1.410 mg m−3. R1 = 0.0740, wR2 (all data) = 0.2379. Goodness of fit on F2 = 1.046. CCDC 1419968, and 1419967 contains the supplementary crystallographic data of 2 and 3, respectively, for this paper.
- (a) M. Nakamura, T. Ikeue, Y. Ohgo, M. Takahashi and M. Takeda, Chem. Commun., 2002, 1198–1199 RSC; (b) S. Kouno, A. Ikezaki, T. Ikeue and M. Nakamura, J. Inorg. Biochem., 2011, 105, 718–721 CrossRef CAS PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- (a) T. Vangberg, R. Lie and A. Ghosh, J. Am. Chem. Soc., 2002, 124, 8122–8130 CrossRef CAS PubMed; (b) H. Hirao, S. Shaik and P. M. Kozlowski, J. Phys. Chem. A, 2006, 110, 6091–6099 CrossRef CAS PubMed; (c) R.-J. Cheng, P.-Y. Chen, T. Lovell, T. Liu, L. Noodleman and D. A. Case, J. Am. Chem. Soc., 2003, 125, 6774–6783 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Instrumentations, experimental details, spectroscopic characterizations, computational details, crystallographic data, DFT-optimized structures, Fig. S1–S13 and Tables S1–S4. CCDC 1419967 (3) and 1419968 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc07111a |
| This journal is © The Royal Society of Chemistry 2015 |