
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 conversion to isocyanate via multiple N–Si bond cleavage at a bulky uranium(III) complex†
Clément
Camp
ab,
Lucile
Chatelain
abc,
Christos E.
Kefalidis
d,
Jacques
Pécaut
ab,
Laurent
Maron
*d and
Marinella
Mazzanti
*c
aUniv. Grenoble Alpes, INAC-SCIB, F-38000 Grenoble, France
bCEA, INAC-SCIB, Reconnaissance Ionique et Chimie de Coordination, 17 Rue des Martyrs, F-38000 Grenoble, France
cInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
dLPCNO, CNRS & INSA, UPS, Université de Toulouse, 135 Avenue de Rangueil, F-31077 Toulouse, France
First published on 8th September 2015
Abstract
The reaction of the sterically saturated uranium(III) tetrasilylamido complex [K(18c6)][U(N(SiMe3)2)4] with CO2 leads to CO2 insertion into the U–N bond affording the stable U(IV) isocyanate complex [K(18c6)][U(N(SiMe3)2)3(NCO)2]n that was crystallographically characterized. DFT studies indicate that the reaction involves the [2+2] cyclo-addition of a double bond of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO to the U–N(SiMe3)2 bond and proceeds to the final product through multiple silyl migration steps.
CO to the U–N(SiMe3)2 bond and proceeds to the final product through multiple silyl migration steps.
The reactivity of uranium(III) with small molecules such as CO2, CO or N2 has been attracting increasing interest in recent years due to the ability of uranium to promote unusual transformations.1,2 Bulky amides have been successfully used in uranium chemistry, as innocent ancillary ligands, as alternatives to the ubiquitous cyclopentadienyl systems.3 In particular the simple neutral [U(N(SiMe3)2)3]4 complex has provided a versatile precursor and has demonstrated interesting reactivity5 including arene reduction and functionalization,6 CO activation,7 and nitride formation.8 In contrast, the ability of the U–N bonds to undergo insertion reactions has been much less explored compared to U–C σ-bonds.1c Only a few examples of insertion of CO2 into U(III)–NR2 and U(IV)–NR2 bonds leading to the formation of U(III)9 and U(IV) carbamates10 have been reported. Examples of the insertion of CO2 into metal–silylamide bonds have been reported11 for main group, d-block and f-block metals12 but are much rarer than the insertion of CO2 into N-alkylamide bonds. In particular, [UIII(N(SiMe3)2)3] was reported to react with CO2 to give O
CNSiMe3 and a second product identified as the tetravalent uranium silanolate [U(OSiMe3)4].12a
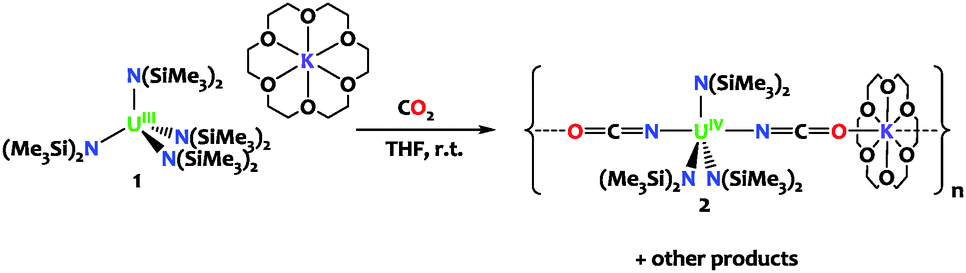
Herein we show that the reaction of CO2 with the sterically saturated uranium(III) tetrasilylamido complex [K(18c6)][U(N(SiMe3)2)4], 1, leads to CO2 insertion into the U–N bond and to the formation of the stable U(IV) isocyanate complex [K(18c6)][U(N(SiMe3)2)3(NCO)2]n2. DFT studies were used in combination with reactivity studies to investigate the mechanism leading to the formation of complex 2. Complex 2 provides a rare example of cyanate formation at a uranium center. To date there have been only three examples of uranium-mediated OCN− formation and they involve the reaction of CO with a nitride,13 imido14 or nitrosyl complex.15
Complex 1 was prepared from [U(N(SiMe3)2)4]16 according to the previously reported procedure.17 The reaction of 1 with 2.5 equivalents of carbon dioxide proceeded slowly (completed after 48 hours) at room temperature affording after workup and recrystallization the U(IV) bis-cyanate complex [K(18c6)][U(N(SiMe3)2)3(NCO)2]n, 2 as a pale pink microcrystalline solid in 48% yield (Scheme 1). The stoichiometry of this reaction requires the presence of additional uranium compounds and reduced by-products (U(III) has been oxidized to U(IV)) that have not been isolated. THF solutions of 2 are stable for at least 48h at room temperature. The 1H NMR spectrum for 2 recorded from deuterated THF at room temperature displays a broad resonance at −10.9 ppm corresponding to the three equivalent {N(SiMe3)2} moieties together with a resonance at 4.7 ppm for the potassium crown ether counter cation.
 | ||
| Scheme 1 | ||
The X-ray crystal structure of 2 shows the presence of a 1D coordination polymer (Fig. 1). The uranium environment is trigonal bipyramidal with three silylamido ligands at equatorial positions and two NCO− anions at apical positions. A K(18c6) cation bridges two NCO ligands from two distinct uranium complexes. The U1–Namido bond distances fall in the range of what was observed in the U(IV) silylamido species. Notably the structure of 2 is closely related to the isoelectronic azide species [Na(THF)4][U{N(SiMe3)2}3(N3)2] reported by Hayton and coworkers5e featuring similar geometry and uranium-ligand bond distances. The three atoms XCX′ units are disordered across the mirror plane of the P21/m space group and refine equally well when N or O or a mixture of both are used at the X and X′ positions. Thus, the X-ray data do not allow us to discriminate between a cyanate (coordination through O) and an isocyanate (coordination through N) ligand. Similar disorder issues were observed in the [{Me2Al(μ-OSiMe3)2Mg(THF)2(μ-OCN)}3] complex11c and in the dimeric U(IV) complex [U(η-C8H6{SiiPr3-1,4}2)(η-Cp*)(NCO)]2.15 However, in most previously reported U(IV) complexes the NCO ligand is N-bound18 and DFT calculations are in agreement with an N-bound coordination (see below and ref. 13). Therefore the structure was refined with N-bound OCN ligands (Fig. 1). The two U–NNCO bond distances at 2.337(3) and 2.338(4) Å are in the range of those found in the few uranium isocyanate complexes reported (2.338(3),13 2.389(6)18b and 2.336(5) Å19).
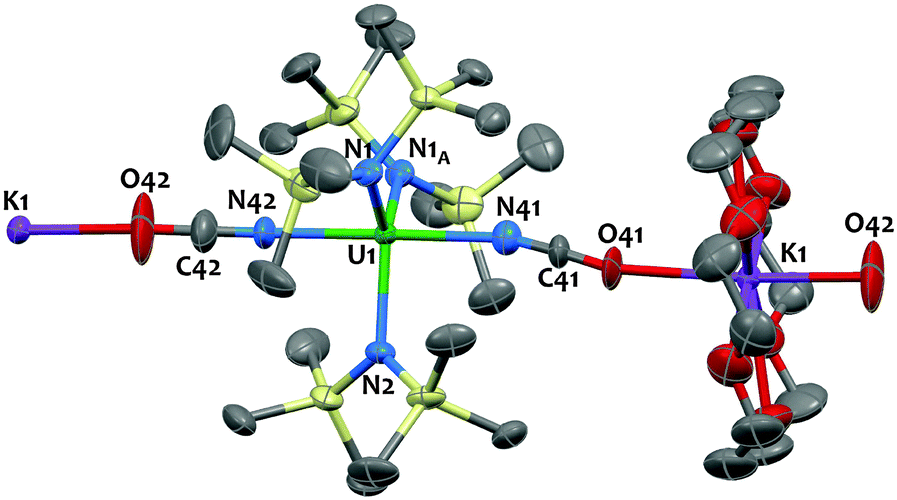 | ||
| Fig. 1 Solid-state molecular structure of [K(18c6)][U(N(SiMe3)2)3(NCO)2]n2 crystallized from THF/hexane. Hydrogen atoms and disorder are omitted for clarity. Uranium (green), potassium (purple), silicon (yellow), nitrogen (blue), oxygen (red) and carbon (grey) atoms are represented with 30% probability ellipsoids. Selected bond distances [Å] and angles [deg]: U1–N1 2.2679(2), U1–N2 2.2696(2), U1–O/N42 = 2.3358(1), U1–O/N41 = 2.3370(1), X42–C42–X′42 = 171.6(1), X41–C41–X′41 = 175.5(1). | ||
The absorption band at 2201 cm−1 in the IR spectrum of 2 was assigned to the asymmetric stretching mode νNCO. This value is similar to those found in the few terminal18 or bridging15 U(IV) and U(III)13 isocyanate complexes reported (2199–2122 cm−1).
All the spectroscopic and analytical data (see ESI†) support the assignment of the three atoms in 2 as NCO ligands. Notably the quaternary carbon (after reaction with 13CO2) resonance at δ = 492.4 ppm in the 13C NMR spectrum, the microanalytical data, and the parent ion in the mass spectrum for the anion [U(N(SiMe3)2)3(NCO)2]− (m/z = 802.4) are in agreement with the assigned formula.
1H and 13C NMR studies of the reaction of 1 with 13CO2 were performed. After addition of 1 equivalent of 13CO2 the reaction leads to the slow disappearance of the 1H signals assigned to complex 1 with completion after 48 hours. The 13C NMR spectrum of the final reaction mixture shows the presence of a signal at 307 ppm that could be assigned to the quaternary carbon of a carbamate or an isocyanate intermediate. No evident color change was observed during the reaction. Additional 13C NMR signals are also observed in the 5.6–1.8 ppm region. Further addition of 1 equivalent of 13CO2 leads to a slow color change of the reaction mixture from dark purple to light pink. 13C NMR monitoring of the reaction showed a slow evolution with time with the disappearance of the 13C NMR signal at 307 ppm and the appearance of the 13C NMR signal at 492.4 ppm assigned to the isocyanate complex 2. All the 13C NMR resonances in the 5.6–1.8 ppm region remained present in the final 13C NMR spectrum but with increased intensity. In particular, two signals at 0.07 ppm in the 1H NMR spectrum and at 1.84 ppm in the 13C NMR spectrum were assigned to the hexamethyldisiloxane (SiMe3)2O by-product. Signals for SiMe3NCO were not observed. The 1H NMR spectrum of the final reaction mixture showed the resonances assigned to complex 2 and additional broad shifted resonances assigned to unidentified U(IV) products. Mass spectrometry studies showed the presence of [U2(OSiMe3)9]− and [U2(NCO)(OSiMe3)8]− species (see ESI†).
In order to shed light on the mechanistic aspects of this peculiar reactivity outcome, DFT investigations using the B3PW91 functional were performed. The system of choice is the full anionic system [U(N(SiMe3)2)4]−, where the counter-cation is not taken into account.13 The anionic [U(IV)–CO2˙]− intermediate Int1 is formed from the electronic reduction of CO2 that occurs at the coordination to the initial U(III) complex (so-called coordination-induced reduction).20 The first step of the mechanism involves the [2+2] cyclo-addition of a double bond from the OCO˙ radical anion to the U–N(SiMe3)2 bond, as it is shown in Fig. 2. The activation barrier for this process is found to be relatively small (14.8 kcal mol−1). In particular, in the transition state the amide group that is involved in the insertion step is considerably far from the uranium center, at a non-bonding distance (dU–N = 3.63 Å). The nature of this late-transition state is most probably due to the directionality of the occupied molecular orbital of the nitrogen with respect to the electrophilic carbon of the carbon dioxide molecule. Moreover, the steric hindrance of the ligand environment may also play an important role in the non-bonding situation between the nitrogen and the uranium, providing a logical explanation for the outcome of this step. In particular, the IRC calculation did not converge into an O,N-bound carbamate intermediate, but instead into an O,O-bound one (Int2), with a subsequent significant drop in enthalpy energy (43.0 kcal mol−1 with respect to the CO2 reduced adduct).
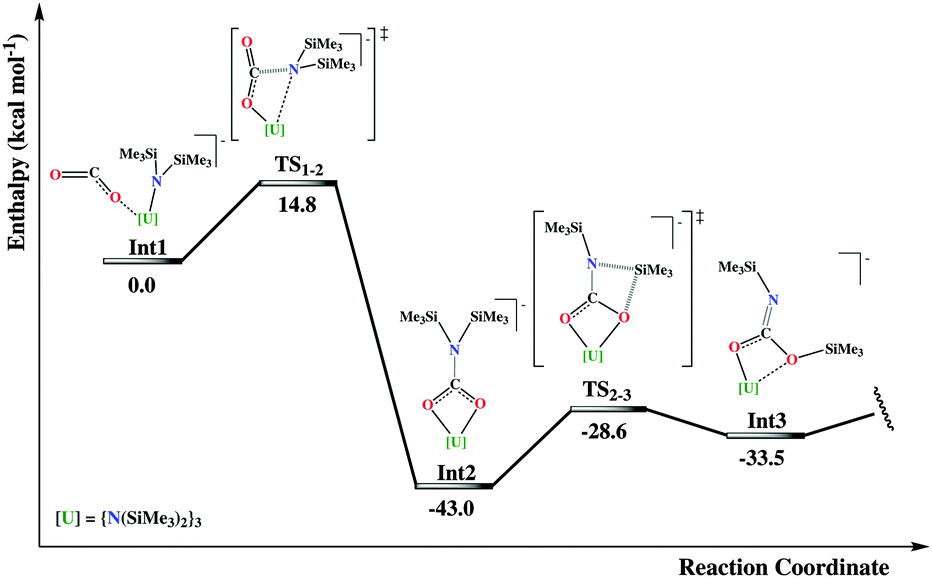 | ||
| Fig. 2 Part of the energy profile that leads to the formation of the isocyanate complex 2. | ||
From the ligated dianionic O,O-bound carbamate the reaction can proceed through a sequence of two successive silyl migration steps (Fig. 2 and 3).
 | ||
| Fig. 3 Part of the energy profile that leads to the formation of the isocyanate complex 2. | ||
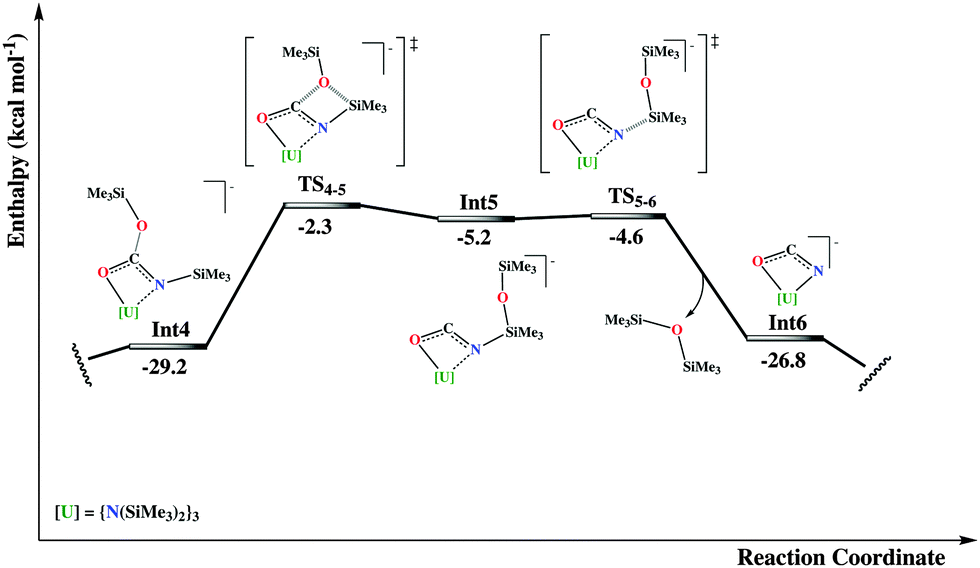
More specifically, the first step refers to the migration of the trimethylsilyl group from the nitrogen atom to one oxygen of the carbamate group, surmounting a moderate activation energy barrier of 14.4 kcal mol−1. From the ensuing intermediate, Int3, rotation around the Osiloxide–C bond of the silyl group is required for the Si atom to approach the coordinated oxygen atom, for the second migration to occur. This isomerization is an almost thermo-neutral process, with enthalpy energy difference of only 4.3 kcal mol−1 in favor of Int3. The following migration of the silyl group is more energy demanding than the previous one, with an accessible activation energy barrier of 26.9 kcal mol−1 in terms of ΔH‡.
In the following intermediate, Int5, a kind of cyanate fragment has been formed, now developing an η2-N,O-type coordination with the uranium atom. This fragment can easily expel a siloxane molecule, which is observed experimentally, through an almost barrierless energy micro-step, viaTS5–6. The resulting intermediate, Int6, is formally described as an OCN2− complex of U(IV). At this point, one can envision two different potential paths, the first corresponding to the isomerization of the cyanate group to give a terminal bonded isocyanate complex or to the nucleophilic attack by a second CO2 molecule on the carbon atom of the OCN2− moiety (Fig. 4). The first possibility results in an important stabilization energy of 21.4 kcal mol−1 with respect to the η2-bound one. Then a putative free NCO radical (originating from a second uranium complex) can coordinate the vacant coordination site of Int7, on the axial position, trans to the other isocyanate, of the trigonal bipyramid. This can be done through two different coordination modes since OCN− is an ambident ligand. Even though both correspond to highly exothermic processes (more than 100 kcal mol−1 stabilization energy), the N-bound intermediate, Int8, is more stable than the O-bound one, (see Int8′ in the ESI†), by almost 10 kcal mol−1. This observation suggests that most probably the two OCN groups in the X-ray structure of complex 2 are bound to the uranium through their nitrogen atoms while the oxygen atoms bind the potassium atoms of the K(18c6) cation.
 | ||
| Fig. 4 Part of the energy profile that leads to the formation of the isocyanate complex 2. | ||
Besides, since the experimental reactivity is found to be influenced by the number of CO2 equivalents reacted with U(III), a second CO2 molecule can undergo a nucleophilic attack at the carbon of the η2-cyanate group in intermediate Int6 (Fig. 4). This will result in the formation of an oxalate-like complex, Int9. Such reactivity is reminiscent of the oxalate formation in U(III) chemistry.21 Two molecules of Int9 can then disproportionate in order to give the bis-cyanate product Int8 and a CO22− complex (Int10). Also in this route, an important stabilization energy is found (almost 130 kcal mol−1). Attempts to compute a reaction pathway involving the insertion of CO2 into a U(III) species were not successful but showed that such a pathway is higher in energy.
In conclusion the bulky uranium(III) tetrasilylamido complex [K(18c6)][U(N(SiMe3)2)4] reacts with CO2 to afford a rare example of a U(IV) isocyanate complex. DFT computational studies suggest that the reaction proceeds through carbon dioxide reduction followed by the [2+2] cyclo-addition of the carbonyl double bond of the reduced carbon dioxide to the U–N(SiMe3)2 bond and multiple silyl migration. The reactivity of this bulky “ate” complex differs from that reported for the neutral analogue [U(N(SiMe3)2)3]12a highlighting the importance of the coordination environment for controlling the CO2 conversion at the uranium center.
This work was supported by the CEA, the Swiss National Science Foundation, and by the EPFL. We thank O. Cooper for recording some analytical data. LM is member of the Institut Universitaire de France. Humboldt Foundation and Calmip are also acknowledged.
Notes and references
-
(a) P. L. Arnold, Chem. Commun., 2011, 47, 9005–9010 RSC
; (b) O. P. Lam and K. Meyer, Polyhedron, 2012, 32, 1–9 CrossRef CAS PubMed
-
(a) A. R. Fox, S. C. Bart, K. Meyer and C. C. Cummins, Nature, 2008, 455, 341–349 CrossRef CAS PubMed
-
(a) D. M. King and S. T. Liddle, Coord. Chem. Rev., 2014, 266, 2–15 CrossRef PubMed
-
(a) R. A. Andersen, Inorg. Chem., 1979, 18, 1507–1509 CrossRef CAS
-
(a) D. L. Clark, M. M. Miller and J. G. Watkin, Inorg. Chem., 1993, 32, 772–774 CrossRef CAS
- P. L. Arnold, S. M. Mansell, L. Maron and D. McKay, Nat. Chem., 2012, 4, 668–674 CrossRef CAS PubMed
- P. L. Arnold, Z. R. Turner, R. M. Bellabarba and R. P. Tooze, Chem. Sci., 2011, 2, 77–79 RSC
- S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888–6889 CrossRef CAS PubMed
- E. M. Matson, P. E. Fanwick and S. C. Bart, Organometallics, 2011, 30, 5753–5762 CrossRef CAS
-
(a) K. W. Bagnall and E. Yanir, J. Inorg. Nucl. Chem., 1974, 36, 777–779 CrossRef CAS
-
(a) W. Sattler and G. Parkin, J. Am. Chem. Soc., 2011, 133, 9708–9711 CrossRef CAS PubMed
-
(a) S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed
- P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 10412–10415 CrossRef CAS PubMed
- I. Castro-Rodriguez and K. Meyer, Chem. Commun., 2006, 1353–1368 RSC
- A. S. P. Frey, F. G. N. Cloke, M. P. Coles and P. B. Hitchcock, Chem. – Eur. J., 2010, 16, 9446–9448 CrossRef CAS PubMed
- A. J. Lewis, U. J. Williams, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2013, 52, 7326–7328 CrossRef CAS PubMed
- L. C. J. Pereira, C. Camp, J. T. Coutinho, L. Chatelain, P. Maldivi, M. Almeida and M. Mazzanti, Inorg. Chem., 2014, 53, 11809–11811 CrossRef CAS PubMed
-
(a) M. J. Crawford, P. Mayer, H. Noth and M. Suter, Inorg. Chem., 2004, 43, 6860–6862 CrossRef CAS PubMed
- I. Castro-Rodriguez, H. Nakai, L. N. Zakharov, A. L. Rheingold and K. Meyer, Science, 2004, 305, 1757–1759 CrossRef CAS PubMed
-
(a) L. Castro, C. E. Kefalidis, D. McKay, S. Essafi, L. Perrin and L. Maron, J. Chem. Soc., Dalton Trans., 2014, 43, 12124–12134 RSC
-
(a) A.-C. Schmidt, F. W. Heinemann, C. E. Kefalidis, L. Maron, P. W. Roesky and K. Meyer, Chem. – Eur. J., 2014, 20, 13501–13506 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, full crystallographic data, 1H and 13C NMR spectra, ESI/MS spectra and computational data are included. CCDC 1417064. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc06707c |
| This journal is © The Royal Society of Chemistry 2015 |