
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Length control of supramolecular polymeric nanofibers based on stacked planar platinum(II) complexes by seeded-growth†
Matthew E.
Robinson
a,
David J.
Lunn
a,
Ali
Nazemi
a,
George R.
Whittell
a,
Luisa
De Cola
b and
Ian
Manners
*a
aSchool of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: ian-manners@bristol.ac.uk
bISIS & icFRC, Université de Strasbourg & CNRS, 8 rue Gaspard Monge, 67000 Strasbourg, France
First published on 10th September 2015
Abstract
The formation of high aspect ratio supramolecular polymeric nanofibers from square-planar platinum(II) complexes through Pt⋯Pt and π–π stacking interactions has been achieved with a small width (<15 nm), tunable length, and relatively narrow length distributions up to ca. 400 nm under conditions of kinetic control using small seed fibers as initiators.
The study of supramolecular polymers, in which directional, non-covalent interactions such as hydrogen-bonding and π-stacking generate the main chain structure from individual monomer units, represents a vibrant interdisciplinary scientific field.1–5 Unlike most covalent polymers which possess “static” structures, supramolecular polymers generally exhibit dynamic behavior.6 This offers opportunities for facile, low viscosity processing and access to stimuli-responsive materials with a variety of applications.1–7 The formation of many supramolecular polymers involves a nucleation–elongation mechanism analogous to a chain-growth covalent polymerization where a slow, unfavourable nucleation step is followed by rapid, favorable elongation.2,4,5 However, a key challenge in the field is to obtain molar mass control and access to monodisperse and segmented (“block”) structures. This is readily achieved for covalent polymers through “living” chain-growth polymerizations, where addition of an initiator leads to chain propagation under conditions where side reactions, such as chain transfer and termination, are absent. Significantly, isolation of monodisperse and segmented supramolecular polymers also requires the use of conditions where intrinsic dynamic behavior is suppressed in order to prevent the rapid equilibration that would lead to molar mass distribution broadening and microstructure randomization.2,8–14
A particularly interesting class of supramolecular polymers is formed by square-planar transition metal complexes typically based on Pt(II) or Au(I)/(III) centres and one or more flexible solvophilic ligands.15–20 These species readily aggregate by cofacial stacking modulated by π–π, metallophilic, and often hydrogen bonding interactions to yield multimicron-long 1D fibers that form organogels. The fibers offer promising emissive properties, resistance to photobleaching, excitation with red or near IR light for bioimaging applications, and potential anticancer activity.15–18 However, although the aggregation behavior can be modified by the nature of the solvophilic ligand(s), control of the growth process is severely limited. Fiber length control is highly desirable as this may allow optoelectronic properties to be tuned and access to long-term colloidal stability to facilitate processing and utility.
Recent work has shown that block copolymers (BCPs) with a crystallizable core-forming block self-assemble in selective solvents for the complementary corona-forming segment to form fiber-like micelles with a distribution of lengths.21–25 The formation of fibers that are polydisperse in length can be attributed to the slow nucleation step preceding elongation.2 This problem can be circumvented by the use of a ‘seeded growth’ method whereby the polydisperse fibers are first cleaved by ultrasound to form very short seed micelles that elongate on the subsequent addition of molecularly dissolved BCP (unimer).26–28 Under conditions of kinetic control, this yields monodisperse micelles with a length determined by the ratio of the amount of added BCP to that of the seed.26 This process has been termed26 “living crystallization-driven self-assembly (CDSA)” due to the analogy with a living covalent polymerization of monomers in which the chain length of the resulting monodisperse polymer is determined by the monomer to initiator ratio.22,26,29,30 This type of seeded-growth approach has recently been successfully applied to the formation of supramolecular 1D polymers formed by Zn porphyrins,31 hexabenzocoronenes,32 perylene diimides,33 and peptide-based macrocycles.34 In this communication we briefly report on our very promising preliminary results that demonstrate a similar method can be applied to the formation of supramolecular nanofibers from planar Pt(II) complexes.
In this work we focused on self-assembly of an analog of the square-planar Pt(II) complexes with short tetra(ethylene glycol) (TEG) pendent groups attached to the metal-bond pyridyl ligand of the type examined by De Cola and coworkers.35 The TEG-containing species were shown to self-assemble into organogels consisting of multimicron long supramolecular fibers of substantial width (ca. 50–100 nm) comprised of multiple π-stacked polymer chains. To facilitate more controlled assembly, we targeted a material with a longer and therefore more spatially demanding oligo(ethylene glycol) (OEG)-functionalized pyridyl solubilizing substituent to limit inter-chain packing at the fiber core. Using this design, we prepared rod-like structures with a small diameter closer to the anticipated diameter for a single supramolecular polymer chain (ca. 1.055 nm based on density functional theory (DFT) calculations for a similar structure, ESI,† Fig. S7).36
The OEG-functionalized pyridyl solubilizing ligand (2) was prepared from tosylated OEG monomethyl ether (1) (Mn = 750 g mol−1) and 4-hydroxypyridine. A mixture of 2, PtCl2(dimethyl sulfoxide)2 as Pt(II) source, and a tridentate N^N^N ligand (2,6-di(1H-tetrazol-5-yl)pyridine) in the presence of an HCl acceptor was refluxed in acetonitrile (MeCN). After purification, 3 was isolated as a yellow solid in 66% yield and characterized by 1H, 13C NMR as well as matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectroscopy (for characterization data see ESI†).
Before conducting the controlled solution self-assembly studies, to gain insight into the stacking behavior of this molecule, UV-visible absorption data of complex 3 in chloroform, CHCl3, at varying concentrations was obtained at room temperature (21 °C) (ESI,† Fig. S6a). At concentrations below 0.1 mg mL−1, the pyridine- and tridentate-ligand-centred (1LC) π–π* transitions were found at 315 and 335 nm, respectively. This is consistent with 3 existing in a molecularly dissolved (unimer) state under these conditions. However, on increasing the concentration to 0.1 mg mL−1 a metal–metal to ligand charge transfer band (1MMLCT), which arises from strong intermolecular interactions between the planar complexes, was observed.37–39 This is consistent with the formation of supramolecular polymeric stacks at concentrations exceeding 0.1 mg mL−1. Having this result in hand, the self-assembly of complex 3 was carried out in solvents selective for the OEG ancillary ligand (Scheme 1).
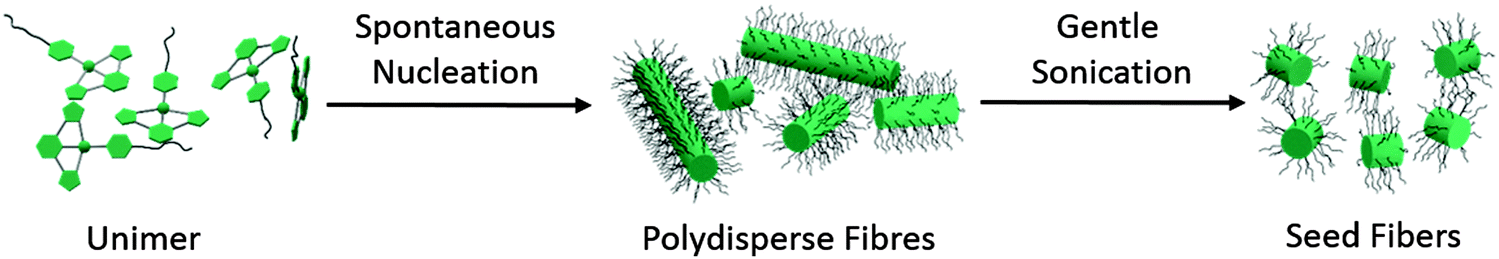 | ||
| Scheme 1 Schematic representation of the formation of fibers from 3. Dissolving the material in an OEG-selective solvent produces a solution of polydisperse length cylinders; sonicating a solution of polydisperse fibers forms short seed fibers. | ||
Dissolution of the solid product 3 in organic solvents such as CHCl3, N,N-dimethylformamide (DMF) and MeCN at concentrations between 0.5–2 mg mL−1 formed clear yellow solutions at 21 °C when left to age over 24 h. Drop-casting these solutions on carbon-coated copper grids followed by solvent evaporation and TEM analysis revealed the formation of polydisperse fibers (ESI,† Fig. S6) (e.g. in CHCl3 – Ln = 206 nm, Lw = 355 nm, Lw/Ln = 1.73, σ/Ln = 0.85 where Ln is the number-average and Lw the weight-average contour length, and σ is the standard deviation). This is typical for an elongation process preceded by spontaneous nucleation (Fig. 1b).
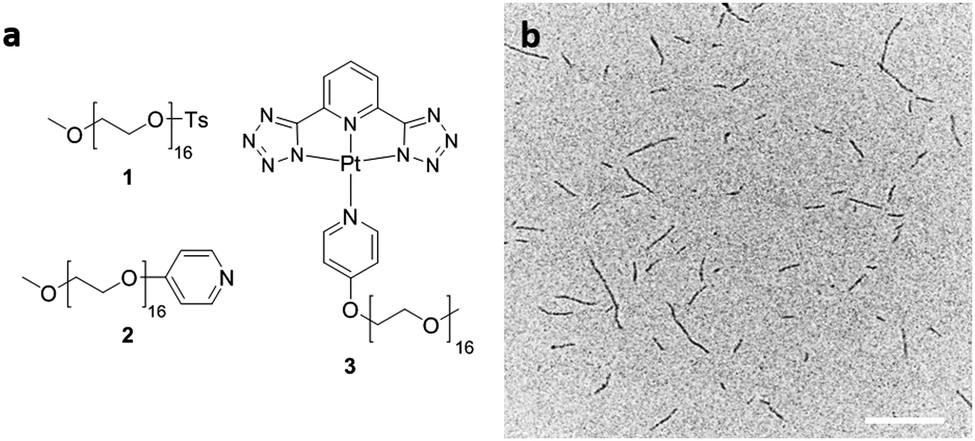 | ||
| Fig. 1 (a) Structures of 1, 2, and 3 and (b) TEM image of polydisperse fibers formed by dissolving 3 in CHCl3 at 0.5 mg mL−1. Scale bar: 500 nm. | ||
Inspection of the TEM images for samples with concentrations between 0.1 and 1 mg mL−1 in CHCl3 and MeCN, indicated that the fibers have small diameters (<15 nm). The smaller diameter of these fibers compared to those discussed earlier for the TEG analogue35 is likely due to the increased length of the solubilizing ligand. Significantly, staining of these nanostructures was unnecessary for TEM imaging as sufficient contrast was observed for the fibers relative to the carbon film as a result of the electron density afforded by the stacked Pt-containing core-forming moiety. Based on TEM analysis of aliquots of the solution in MeCN, the fibers could be returned to their unimer state by heating the solution to 80 °C for 1 h. When the solution was cooled to room temperature over 12 h, cylinders reformed slowly and patches of a film, presumably formed by unimer, were present on the TEM images (ESI,† Fig. S8).
We explored the use of a seeded growth approach based on the living CDSA method developed for BCPs with a crystallizable core-forming block. First, small seed fibers were prepared in chloroform. This was achieved by subjecting polydisperse fibers of 3, prepared in CHCl3, to gentle sonication at 0 °C for 1 h in a sonication bath. The average contour length of the resulting fibers was determined by TEM analysis (Ln = 60 nm, Lw = 68 nm, Lw/Ln = 1.14, σ/Ln = 0.35) (ESI,† Fig. S9).
The ability to control fiber length was then examined through the addition of unimers of 3 in MeCN to the seeds. To separate solutions containing 20 μg of seed fibers in CHCl3 (200 μL, 0.1 mg mL−1) at 21 °C, different amounts of unimer (5 μg, 10 μg, 20 μg, 40 μg, and 80 μg) were added from an MeCN solution (2 mg mL−1) heated to 80 °C. The solutions were aged for 24 h at 21 °C. TEM analysis showed the lengths (Ln) of the fibers had increased to 150, 178, 254, 390, and 602 nm, respectively. The length distributions were narrow up to Ln = 390 nm (Lw/Ln = 1.21) but increased for the longest fibers (Fig. 2 and Table 1).
 | ||
| Fig. 2 (a) Schematic representation of the formation of elongated fibers. (b) Linear dependence of average contour length (Ln) on the unimer to seed mass ratio. (c) Seed fibers (Ln = 60 nm, Lw/Ln = 1.14) formed through gentle sonication of polydisperse fibers (d–f) TEM micrographs of elongated fibers obtained by adding (d) 20 μg, (e) 40 μg, and (f) 80 μg of unimer (as a 2 mg mL−1 solution in hot MeCN) to 20 μg of seeds. Scale bars: 250 nm. | ||
| Length data | Seeds | m unimer/mseed | ||||
|---|---|---|---|---|---|---|
| 0.25 | 0.5 | 1 | 2 | 4 | ||
| L n (nm) | 60 | 150 | 178 | 254 | 390 | 602 |
| L w (nm) | 68 | 182 | 236 | 311 | 473 | 934 |
| L w/Ln | 1.14 | 1.22 | 1.33 | 1.22 | 1.21 | 1.55 |
| σ/Ln | 0.37 | 0.45 | 0.57 | 0.47 | 0.46 | 0.47 |
Interestingly, the contour length of the resulting fibers is larger than that predicted based on the unimer to seed mass ratio. This is particularly evident after the first addition to the seeds (Ln = 60 nm) where the resulting fibers were found to possess a length of Ln = 150 nm (Fig. 2b). A significantly larger than expected increase in average contour length has also been observed in other systems and may be due to fiber width of the elongated section being smaller than that of the seed.40,41 It is also possible that, in the case of the first addition, some of the seeds formed by sonication are not active to further growth. The increase in the breadth of the length distribution for fibers of length Ln = 600 nm may be a consequence of increased competition from spontaneous nucleation when larger amounts of unimer are added. We also examined the UV-vis absorption spectra for fibers of differing length and observed no shift in the MMLCT band (ESI,† Fig. S15). This suggests that the maximum conjugation length is <50 nm.
The supramolecular fibers formed by 3 have the potential to undergo dynamic exchange of the monomeric Pt(II) building blocks. To explore this possibility, the length distribution for a sample in chloroform at 0.1 mg mL−1 with Ln = 247 nm, Lw/Ln = 1.30 was monitored over the course of 3 days. However, no appreciable difference in Ln or Lw/Ln was detected within experimental error, which suggests that the fibers are kinetically trapped under the conditions used (ESI,† Fig. S16).
The preliminary results demonstrate that the use of a seeded growth approach, analogous to the living CDSA method used to control the length of fiber-like BCP micelles, can be applied to a planar Pt(II) complex with pendent OEG substituents (3) that forms supramolecular polymeric fibers. Rather than forming the gels characteristic of long fibers generated in previous work under uncontrolled growth conditions involving spontaneous nucleation, the shorter (up to ca. 600 nm) fibers formed by 3 possess relatively narrow length distributions (especially up to ca. 400 nm). The fibers formed remain colloidally stable in solution for at least 6 months (ESI,† Fig. S17). Furthermore, no significant change in the fiber lengths or length distributions was noted over 3 days indicating that the seeded growth process proceeds under kinetic control where dynamic exchange processes are effectively suppressed.
In summary, these promising preliminary results with supramolecular polymers based on planar Pt(II) complexes suggest that the seeded growth approach is likely to be applicable to a wide variety of planar species based on different metal/ligand combinations that are known to stack to form fiber-like structures and also to other related systems. Moreover, the addition of BCPs with different corona- or core-forming blocks to seed micelles has been shown to yield well-defined block co-micelles with segmented structures by living CDSA. A similar approach applied to self-assembling planar metal complexes would be expected to give rise to unique segmented structures with interesting combinations of properties and potential for hierarchical assembly.42 The preparation of more well-defined and much shorter seed fibers26 in order to further improve growth uniformity and the polydispersity of the fiber samples is also an important future target.
We thank the University of Bristol for funding this preliminary work.
Notes and references
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- C. Rest, R. Kandanelli and G. Fernandez, Chem. Soc. Rev., 2015, 44, 2543–2572 RSC.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed.
- L. Albertazzi, D. van der Zwaag, C. M. A. Leenders, R. Fitzner, R. W. van der Hofstad and E. W. Meijer, Science, 2014, 344, 491–495 CrossRef CAS PubMed.
- R. D. Mukhopadhyay and A. Ajayaghosh, Science, 2015, 349, 241–242 CrossRef CAS PubMed.
- J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS PubMed.
- D. van der Zwaag, T. F. A. de Greef and E. W. Meijer, Angew. Chem., Int. Ed., 2015, 54, 8334–8336 CrossRef CAS PubMed.
- D. J. Lunn, J. R. Finnegan and I. Manners, Chem. Sci., 2015, 6, 3663–3673 RSC.
- Z. Huang, L. Yang, Y. Liu, Z. Wang, O. A. Scherman and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 5351–5355 CrossRef CAS PubMed.
- Q. Zhang and H. Tian, Angew. Chem., Int. Ed., 2014, 53, 10582–10584 CrossRef CAS PubMed.
- J. Kumar, H. Tsumatori, J. Yuasa, T. Kawai and T. Nakashima, Angew. Chem., Int. Ed., 2015, 54, 5943–5947 CrossRef CAS PubMed.
- G. Odian, Principles of Polymerization, John Wiley & Sons, Inc., New Jersey, 2004 Search PubMed.
- J.-J. Zhang, W. Lu, R. W.-Y. Sun and C.-M. Che, Angew. Chem., Int. Ed., 2012, 51, 4882–4886 CrossRef CAS PubMed.
- X.-S. Xiao, W.-L. Kwong, X. Guan, C. Yang, W. Lu and C.-M. Che, Chem. – Eur. J., 2013, 19, 9457–9462 CrossRef CAS PubMed.
- M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144–4166 RSC.
- V. K.-M. Au, D. Wu and V. W.-W. Yam, J. Am. Chem. Soc., 2015, 137, 4654–4657 CrossRef CAS PubMed.
- V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506–6507 CrossRef CAS PubMed.
- C. A. Strassert, C.-H. Chien, M. D. Galvez Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946–950 CrossRef CAS PubMed.
- J. A. Massey, K. Temple, L. Cao, Y. Rharbi, J. Raez, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2000, 122, 11577–11584 CrossRef CAS.
- N. Petzetakis, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2011, 2, 955–960 RSC.
- Z.-X. Du, J.-T. Xu and Z.-Q. Fan, Macromolecules, 2007, 40, 7633–7637 CrossRef CAS.
- H. Schmalz, J. Schmelz, M. Drechsler, J. Yuan, A. Walther, K. Schweimer and A. M. Mihut, Macromolecules, 2008, 41, 3235–3242 CrossRef CAS.
- M. Lazzari, D. Scalarone, C. E. Hoppe, C. Vazquez-Vazquez and M. A. Lòpez-Quintela, Chem. Mater., 2007, 19, 5818–5820 CrossRef CAS.
- J. B. Gilroy, T. Gädt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nature Chem., 2010, 2, 566–570 CrossRef CAS PubMed.
- T. Gadt, N. S. Ieong, G. Cambridge, M. A. Winnik and I. Manners, Nature Mater., 2009, 8, 144–150 CrossRef PubMed.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed.
- J. Qian, X. Li, D. J. Lunn, J. Gwyther, Z. M. Hudson, E. Kynaston, P. A. Rupar, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2014, 136, 4121–4124 CrossRef CAS PubMed.
- J. Schmelz, A. E. Schedl, C. Steinlein, I. Manners and H. Schmalz, J. Am. Chem. Soc., 2012, 134, 14217–14225 CrossRef CAS PubMed.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nature Chem., 2014, 6, 188–195 CrossRef CAS PubMed.
- W. Zhang, W. Jin, T. Fukushima, A. Saeki, S. Seki and T. Aida, Science, 2011, 334, 340–343 CrossRef CAS PubMed.
- (a) L. Bu, T. J. Dawson and R. C. Hayward, ACS Nano, 2015, 9, 1878–1885 CrossRef CAS PubMed; (b) S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS PubMed.
- A. Pal, M. Malakoutikhah, G. Leonetti, M. Tezcan, M. Colomb-Delsuc, V. D. Nguyen, J. van der Gucht and S. Otto, Angew. Chem., Int. Ed., 2015, 54, 7852–7856 CrossRef CAS PubMed.
- N. K. Allampally, C. A. Strassert and L. De Cola, Dalton Trans., 2012, 41, 13132–13137 RSC.
- M. Mauro, A. Aliprandi, C. Cebrian, D. Wang, C. Kubel and L. De Cola, Chem. Commun., 2014, 50, 7269–7272 RSC.
- M. Mydlak, M. Mauro, F. Polo, M. Felicetti, J. Leonhardt, G. Diener, L. De Cola and C. A. Strassert, Chem. Mater., 2011, 23, 3659–3667 CrossRef CAS.
- C. A. Strassert, M. Mauro and L. De Cola, Adv. Inorg. Chem., 2011, 63, 47–103 CrossRef CAS PubMed.
- J. A. G. Williams, in Photochemistry and Photophysics of Coordination Compounds II, ed. V. Balzani and S. Campagna, Springer, Berlin Heidelberg, 2007 Search PubMed.
- J. Gwyther, J. B. Gilroy, P. A. Rupar, D. J. Lunn, E. Kynaston, S. K. Patra, G. R. Whittell, M. A. Winnik and I. Manners, Chem. – Eur. J., 2013, 19, 9186–9197 CrossRef CAS PubMed.
- H. Qiu, Y. Gao, V. A. Du, R. Harniman, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2015, 137, 2375–2385 CrossRef CAS PubMed.
- H. Qiu, Z. M. Hudson, M. A. Winnik and I. Manners, Science, 2015, 347, 1329–1332 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, method of self-assembly and length measurement data for seeded growth trials. See DOI: 10.1039/c5cc06606a |
| This journal is © The Royal Society of Chemistry 2015 |