
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sonochemical degradation of N-methylpyrrolidone and its influence on single walled carbon nanotube dispersion†
Hin Chun
Yau
a,
Mustafa K.
Bayazit
b,
Joachim H. G.
Steinke
a and
Milo S. P.
Shaffer
*a
aDepartment of Chemistry, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK. E-mail: m.shaffer@imperial.ac.uk
bDepartment of Chemical Engineering, University College London, London, WC1E 7JE, UK
First published on 15th September 2015
Abstract
Sonicating pure N-methyl pyrrolidone (NMP) rapidly produces contaminating organic nanoparticles, at increasing concentration with time, as investigated by AFM, as well as UV-vis, IR and NMR spectroscopies. The contamination issue affects carbon nanotube, and likely other nanomaterial, dispersions processed by sonication in organic solvents.
Single walled carbon nanotubes (SWNTs) are widely incorporated into applications such as thin-film transistors (TFTs), transparent conductors and composites.1 In general, to utilise their full potential, dispersions of highly individualised, clean SWNTs are often desired, without contaminating amorphous carbon, graphitic particles or catalyst. There are a number of well-established methods to exfoliate SWNTs including acid oxidation,2 ultrasonication3 and (electro)chemical charging.4 Acid oxidation is the most destructive method as it introduces defects onto SWNTs side wall which have a negative impact on both electronic and mechanical properties of SWNTs. (Electro)chemical charging gives the highest degree of individualised SWNTs, but has to be carried out in inert atmosphere (ideally in a glove box) to avoid side reactions. Ultrasonication is the fastest and simplest dispersion route and does not require dry or inert atmosphere; it can be performed using either a probe (also known as tip or horn) or a sonication bath.3,5 SWNTs can be dispersed in water with the aid of amphiphilic surfactants or macromolecules,3b,6 or in organic, typically amidic solvents, particularly N-methyl pyrrolidone (NMP) (see below), in which no additional surfactants are required.5,7 The successful solvation of SWNTs has been previously attributed to the similar surface energies between the amides and SWNTs.8 However, there are solvents with similar surface energies to amides that do not solvate SWNTs. Clearly, there is at least an extra parameter that needs to be considered together with the surface energy theory when choosing a solvent for SWNT dispersion. One interesting example of a non-amidic solvent for SWNT dispersion is ortho-dichlorobenzene (ODCB) which was reported to give individualised SWNTs in good yield.9 However, further study showed that the stabilisation of SWNTs in ODCB resulted from in situ polymer formation via a sonochemical degradation of ODCB; the oligomers/polymers either adhere or are radically-grafted onto the SWNTs, enhancing solvation of SWNTs by the remaining pristine ODCB.10 Although such sonochemical solvent effects are less widely discussed in the field, it is well recognised that the strong shear force created by sonication is sufficient to introduce defects and shorten SWNTs,11 degrading their electrical conductivity and other properties of interest.12
Atomic force microscopy (AFM) is a classic technique for characterising SWNTs and provides vital information on their diameter, length and degree of individualisation. When studying the dispersion quality using AFM, attention usually focuses only on the linear features associated with the SWNTs. Other non-SWNTs species are often simply assigned as impurities, such as amorphous carbon or catalyst particles inherited from SWNTs synthesis. However, this paper studies their origin in detail, to explore issues associated with sonication in organic solvent. In general, the findings have broad relevance, as the sonication approach has been widely applied and extended to many other nanomaterials including graphene and transition metal dichalcogenide layer materials.13
In order to isolate predominantly individualised SWNTs, dispersions are normally centrifuged at high speed. Due to the difference in density, the majority of large SWNT bundles and catalyst particles are found in the sediment; the supernatant consists mostly of individual SWNTs, small SWNT bundles and amorphous carbon. However, small particulates (<10 nm) are often observed in literature AFM micrographs for centrifuged SWNT samples, particularly for those sonicated in N-methyl pyrrolidone (NMP) (see ESI,† Fig. S1 for an example). A possible rationale for these particulates is the presence of catalyst particles. However, the dense metal catalyst should sediment during centrifugation. To study these particulates in more detail, pre-centrifuged and post-centrifuged SWNT–NMP dispersions were directly compared. HiPco SWNTs were chosen for the study due to their popularity in nanotube research and availability commercially. NMP was again chosen for its popularity and its reported ability to disperse high concentrations8b of individualised SWNTs (116 μg mL−1). The SWNT/NMP mixture was sonicated for 2 hours to achieve a maximum concentration of individualised SWNTs.12
By AFM, the pre-centrifuged SWNT dispersions (Fig. 1a) consisted mostly of aggregated SWNTs and a large quantity of small particulates, with a size distribution from 2 to 15 nm (Fig. 1b). The particulates are distributed uniformly throughout the sample rather than associated with SWNTs, as might be expected for catalyst particles.14 When the same SWNTs dispersion was centrifuged at 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 2 hours, mostly individualised SWNTs and small number of particulates were found in the top 50% of supernatant (Fig. 1c).
000g for 2 hours, mostly individualised SWNTs and small number of particulates were found in the top 50% of supernatant (Fig. 1c).
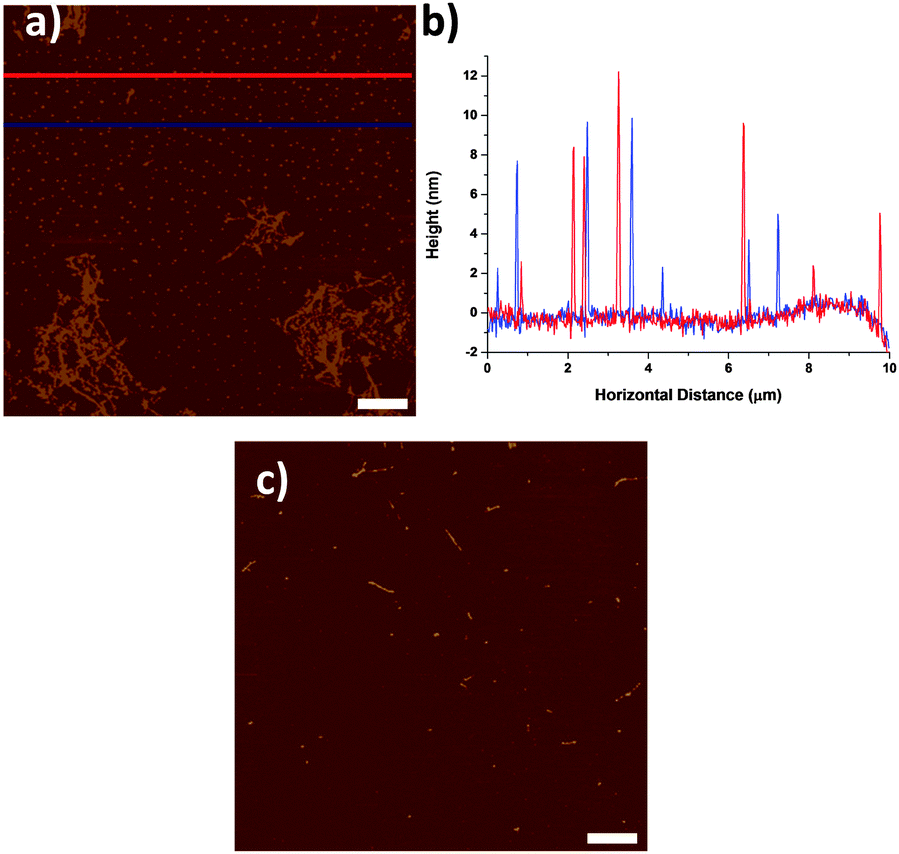 | ||
| Fig. 1 (a) AFM micrograph of HiPco SWNTs dispersed in NMP with 2 hours sonication, before centrifugation. (b) Line profiles of the blue and red lines in (a). (c) AFM micrograph of HiPco SWNTs dispersed in NMP with 2 hours sonication and centrifuged at 200000g for 2 hours. Scale bars are 1 μm. | ||
To establish whether the small particulates might originate from solvent/processing contamination or as a result of a sonochemical reaction, the behavior of pure NMP was examined after sonicating for 0, 5, 30 and 120 min with constant power (150 W). The short times are typical durations for SWNTs dispersion, whereas 120 min was chosen to give a strong signature of any sonochemical effect.5,8a Non-sonicated NMP (Fig. 2a, 0 min sonication) was treated in an identical way as the normal dispersion preparation to provide a reliable control: the sonication probe was immersed into NMP for 120 min (without sonication), whilst cooled with an ice bath. AFM examination (Fig. 2a) clearly showed that the 0 min NMP was relatively clean with very few features, most likely dust particles which disappear after centrifugation (Fig. 2e); it can be concluded that the particulates are not present in the original solvent, and do not arise from handling contamination.
 | ||
| Fig. 2 AFM micrographs of as-sonicated NMP prior centrifugation (a) 0 min, (b) 5 min, (c) 30 min, (d) 120 min; and after centrifugation (e) 0 min, (f) 5 min, (g) 30 min and (h) 120 min. All scale bars are 1 μm. | ||
As the sonication time increased (from 5 to 120 min), the NMP became increasingly yellow (see ESI,† Fig. S2); the number of particulates observed dried on AFM substrates also increased (Fig. 2b–d). It is clear that the particulates originate solely from the effect of sonication on the solvent, as no SWNTs were present. When the as-sonicated NMPs were centrifuged, the majority of the particulates were removed (Fig. 2f–h). However, the pattern of remaining particulates for the 120 min sonicated NMP (Fig. 2d and h) was similar to that in the SWNT–NMP dispersion (Fig. 1a and c), suggesting a similar origin. There are two possible mechanisms which could generate the particulates, either they are generated in situ by a sonochemical degradation of the solvent or they are metal or metal oxide nanoparticles that sheared off from the sonication probe (titanium alloy, Ti-6Al-4V). To identify the particulates, they were isolated from the solvent for analysis. In order to minimise any thermal degradation of the NMP, the remaining solvent was removed from the 120 min sonicated NMP sample by purging under a stream of nitrogen at room temperature. Since NMP has a high boiling point (bp. 202 °C) and low vapour pressure (0.29 mmHg @ 20 °C), it took 2 weeks to evaporate 10 mL of NMP (to constant weight) to give a yellow waxy residue (4 mg, 0.4 wt%). A control experiment of drying as-received NMP similarly with nitrogen produced no measurable residue.
Thermogravimetric analysis (Fig. S3, ESI†) of the yellow residue showed that >95% of the material combusts below 850 °C (under air), excluding metal or metal oxides as a primary constituent. The IR spectrum of the yellow residue (Fig. 3a, red line) shows a medium intensity stretch at 3100–3500 cm−1; corresponding to OH or NH, whereas the NMP IR spectrum (Fig. 3a, black line) shows only a weak feature at around 3500 cm−1, most likely for absorbed moisture. The carbonyl stretch region of the yellow residue (1657 cm−1) is red-shifted compared to the as-received NMP carbonyl stretch (1675 cm−1). The shift in the carbonyl stretching is consistent with a ring opening reaction of the NMP 5-membered ring resulting in a non-cyclic (less strained, red-shifted) amide carbonyl or simple alkyl carbonyl. Hence, the particulates on the AFM substrate are likely to be the product of a sonochemical reaction of NMP. Since the sonication was carried out with an ice bath, water vapour could condense into the sonicating NMP and the localised high temperature generated during sonication (particularly near cavitation bubbles) might induce amide hydrolysis. However, a control experiment with anhydrous NMP (see ESI,† Fig. S4) showed a similar colour change (from colourless to yellow) after 120 min of sonication under nitrogen. It is therefore unlikely that water or oxygen play a significant role in the degradation process. NMP is known to be easily oxidized or degraded at elevated temperature.15 However, no significant bulk solution temperature increase above room temperature (Fig. S5, ESI†) was observed in the current experiment (probe sonication), suggesting that the ice bath was sufficient to dissipate the heat generated. Hence, the observed NMP ring opening and polymerisation can be solely attributed to a sonochemical process. The 1H NMR spectrum of the yellow residue did not show any signals exactly corresponding to pristine NMP (ESI,† Fig. S6) implying the NMP chemical structure was altered and the removal of molecular NMP was successful. The NMP degradation process is likely to be complex, involving a range of mostly radical-based reaction intermediates and subsequent polymerisation, to form a heterogeneous crosslinked oligo/polymer. It is difficult to assign the 1H NMR spectrum fully; however, characteristic features appear in the aldehyde (10.45 ppm) and alkene (5.0–5.5 ppm) regions, consistent with ring opening of NMP and subsequent rearrangement. A possible initial degradation pathway and key structural features of the polymer are discussed further in ESI,† Fig. S6.
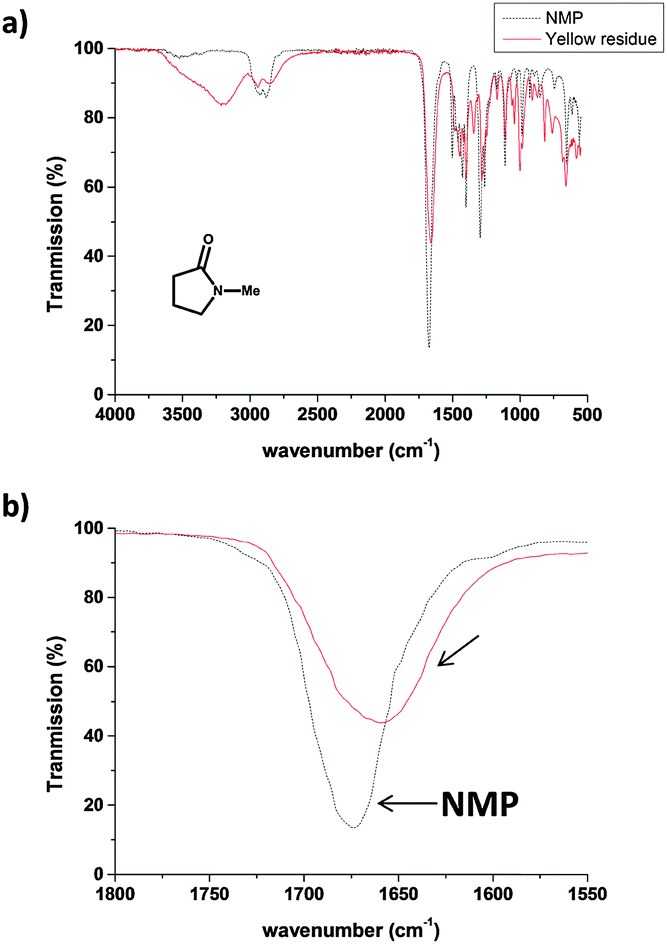 | ||
| Fig. 3 (a) IR spectra of NMP (black dotted line) and the yellow residue (red solid line) isolated from evaporating solvent from the 120 min sonicated NMP. (b) Expanded carbonyl region for the two samples. | ||
When sonicating SWNTs in NMP, the sonochemically degraded NMP may bind covalently to the SWNTs through radical trapping;16 alternatively, oligomerised species may absorb onto the SWNT surface. Either way, a buffer layer bound to the SWNT's surface may affect the chemical and electronic properties. In order to quantify how much degraded NMP binds to the SWNTs, a HiPco dispersion (2 mg SWNTs in 20 mL NMP, 120 min sonicated) was filtered through a PTFE membrane (100 nm pore size) and washed with ethanol to remove excess NMP. The resulting SWNT bucky paper was then dried in vacuo at room temperature overnight to remove ethanol and any unreacted NMP (until constant weight). Thermogravimetric analysis (TGA) under nitrogen quantified the organic residue remaining on the SWNTs which decomposed between 300–500 °C. At 850 °C, only 30% of the original mass remained, indicating that as much as 70 wt% of the buckypaper consisted of an organic residue which could not be removed by ethanol washing. TGA of as-received HiPco SWNTs (under nitrogen) only showed 10% weight loss over the same temperature range (200–850 °C). A control of SWNTs soaked but not sonicated in NMP, followed by identical ethanol washing, showed a similar TGA profile to the pristine SWNTs (Fig. 4). Therefore, estimations of the as-sonicated dispersed SWNT concentration based on weight may be misleading.
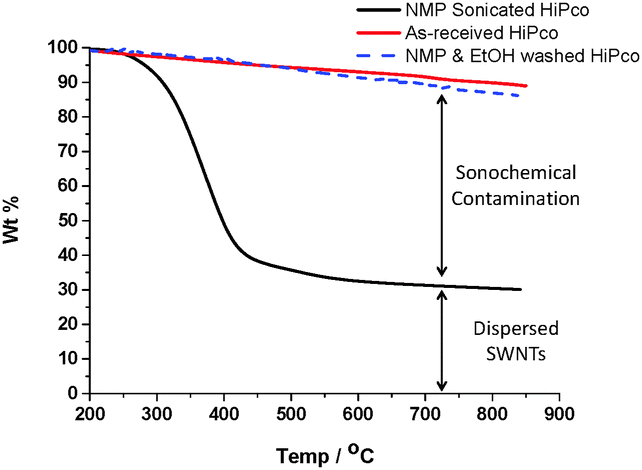 | ||
| Fig. 4 TGA (under nitrogen) of as-received HiPco, a SWNT buckypaper, prepared by sonicating HiPco SWNTs in NMP for 120 min, filtering & washing with ethanol, and HiPco SWNTs stirred in NMP followed by similar ethanol washing. | ||
UV-vis spectra of NMP sonicated for 5, 30 and 120 min showed significant absorbance at around 350 nm. The absorbance likely corresponds to scattering from the nano-particulates in the as-sonicated NMP. In addition to the scattering profile, the absorbance could be also assigned to the optical absorbance of a yellow oligo/polymeric product. Typically, the concentration of the SWNT dispersions is derived from the optical absorbance8a at 660 nm. In order to calculate the true optical absorbance of SWNTs, a solvent background is typically subtracted from the spectrum. In general, a clean (non-sonicated) solvent background is applied. However, as shown in Fig. S7 and S8 (ESI†), the optical absorbances at 660 nm of pristine and sonicated NMP are different. Hence, when a clean solvent background is applied in the calculation of SWNT concentration, a small (though not necessarily negligible) error will be introduced. Since there are many variables in a typical sonication experiment such as time, power, volume of solvent, tip size and cooling medium, it is difficult to standardise a possible error for the established extinction coefficient (3264 mL mg−1 m−1) of SWNTs in NMP. The literature value was derived from a systematic study of a range of SWNT NMP dispersions with known initial concentration and a clean NMP solvent background. In the 120 min example, the corrected SWNT concentration is 16.5 μg mL−1 (with 120 min sonicated NMP as baseline) instead of 17 μg mL−1 (with pristine NMP as baseline) which gives an error of ∼3% in the calculation. Larger errors may occur in other systems, or when applying the extinction coefficient under other circumstances.
Experimental data suggest that NMP degrades and polymerises during sonication, producing contamination by discrete organic nano-particulates (2–15 nm) and some fraction which may bind/adhere to SWNT surface. The sonication times explored covered typical SWNT dispersion protocols; at the longest time tested (120 min), around 70 wt% of the suspended ‘SWNT’ mass fraction is organic contaminant. Even at short sonication times, the contamination may be significant. The presence of this organic material should be considered in subsequent application and for mass determination of concentration. Although centrifugation is generally applied to NMP SWNT dispersions, it does not completely remove the particulates. However, TGA confirmed that the organic nano-particulates burn off at elevated temperature (850 °C, nitrogen). Hence, in order to improve AFM image quality, samples can be annealed in vacuo at elevated temperature to remove the organic nano-particulates. Various degradation pathways of NMP are known, but titania catalysed ring-opening17 of NMP during sonication may be a possible explanation, as water hydrolysis, oxidation and thermal degradation have been ruled out by control experiments. In the presence of SWNTs, it is possible that trace acid, either from carboxylates generated during synthesis or earlier acid purification may play a role in stablising amide dispersions,18 but the pure solvent degrades in any case. The ring opening reaction most likely results in a range of radical-based and other intermediates,17 hence, the subsequent polymer may be cross linked. Identifying the actual polymer structure is challenging but may be useful in understanding the stability of SWNTs in NMP. Since the sonochemically degraded NMP is likely to either adhere or graft covalently onto SWNT surfaces, it may act as a steric barrier layer in an analogous manner to systems including an explicit dispersant, published previously, such as polyvinylpyrrolidone (PVP)/SWNTs/NMP.19 Other amides, such as dimethylformamide (DMF) may behave similarly. Together with the surface energy theory, the idea of steric stability induced by sonochemical degradation products may further our understanding of SWNT dispersion stability. Similar conclusions are likely be applicable to graphene and related 2D materials dispersed by sonication in NMP,13 since the degradation is intrinsic to the solvent. Graphene shares a similar sp2 carbon framework chemistry to SWNTs, and the oligomerized NMP may also graft or adhere to graphene surfaces, contributing to enhanced dispersibility and altered properties.
The authors would like to thank EPSRC (EP/G007314/1) for the support of this research. MKB is grateful to the Scientific and Technological Research Council of Turkey (TUBITAK) for post-doctoral research permission.
Notes and references
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- J. Zhang, H. L. Zou, Q. Qing, Y. L. Yang, Q. W. Li, Z. F. Liu, X. Y. Guo and Z. L. Du, J. Phys. Chem. B, 2003, 107, 3712–3718 CrossRef CAS.
- (a) M. Zheng, A. Jagota, E. D. Semke, B. A. Diner, R. S. McLean, S. R. Lustig, R. E. Richardson and N. G. Tassi, Nat. Mater., 2003, 2, 338–342 CrossRef CAS PubMed; (b) V. C. Moore, M. S. Strano, E. H. Haroz, R. H. Hauge, R. E. Smalley, J. Schmidt and Y. Talmon, Nano Lett., 2003, 3, 1379–1382 CrossRef CAS.
- (a) S. A. Hodge, M. K. Bayazit, H. H. Tay and M. S. P. Shaffer, Nat. Commun., 2013, 4 Search PubMed; (b) S. Fogden, C. A. Howard, R. K. Heenan, N. T. Skipper and M. S. P. Shaffer, ACS Nano, 2011, 6, 54–62 CrossRef PubMed.
- C. A. Furtado, U. J. Kim, H. R. Gutierrez, L. Pan, E. C. Dickey and P. C. Eklund, J. Am. Chem. Soc., 2004, 126, 6095–6105 CrossRef CAS PubMed.
- (a) W. Wenseleers, I. I. Vlasov, E. Goovaerts, E. D. Obraztsova, A. S. Lobach and A. Bouwen, Adv. Funct. Mater., 2004, 14, 1105–1112 CrossRef CAS PubMed; (b) M. F. Islam, E. Rojas, D. M. Bergey, A. T. Johnson and A. G. Yodh, Nano Lett., 2003, 3, 269–273 CrossRef CAS.
- (a) K. D. Ausman, R. Piner, O. Lourie, R. S. Ruoff and M. Korobov, J. Phys. Chem. B, 2000, 104, 8911–8915 CrossRef CAS; (b) J. Liu, M. J. Casavant, M. Cox, D. A. Walters, P. Boul, W. Lu, A. J. Rimberg, K. A. Smith, D. T. Colbert and R. E. Smalley, Chem. Phys. Lett., 1999, 303, 125–129 CrossRef CAS.
- (a) S. Giordani, S. D. Bergin, V. Nicolosi, S. Lebedkin, M. M. Kappes, W. J. Blau and J. N. Coleman, J. Phys. Chem. B, 2006, 110, 15708–15718 CrossRef CAS PubMed; (b) S. D. Bergin, Z. Sun, D. Rickard, P. V. Streich, J. P. Hamilton and J. N. Coleman, ACS Nano, 2009, 3, 2340–2350 CrossRef CAS PubMed.
- (a) Q. Cheng, S. Debnath, E. Gregan and H. J. Byrne, Phys. Status Solidi B, 2008, 245, 1947–1950 CrossRef CAS PubMed; (b) J. L. Bahr, E. T. Mickelson, M. J. Bronikowski, R. E. Smalley and J. M. Tour, Chem. Commun., 2001, 193–194 RSC.
- S. Niyogi, M. A. Hamon, D. E. Perea, C. B. Kang, B. Zhao, S. K. Pal, A. E. Wyant, M. E. Itkis and R. C. Haddon, J. Phys. Chem. B, 2003, 107, 8799–8804 CrossRef CAS.
- G. Pagani, M. J. Green, P. Poulin and M. Pasquali, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11599–11604 CrossRef CAS PubMed.
- S. N. Barman, M. C. LeMieux, J. Baek, R. Rivera and Z. Bao, ACS Appl. Mater. Interfaces, 2010, 2, 2672–2678 CAS.
- (a) G. Cunningham, M. Lotya, C. S. Cucinotta, S. Sanvito, S. D. Bergin, R. Menzel, M. S. P. Shaffer and J. N. Coleman, ACS Nano, 2012, 6, 3468–3480 CrossRef CAS PubMed; (b) Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- I. W. Chiang, B. E. Brinson, A. Y. Huang, P. A. Willis, M. J. Bronikowski, J. L. Margrave, R. E. Smalley and R. H. Hauge, J. Phys. Chem. B, 2001, 105, 8297–8301 CrossRef CAS.
- (a) L. Poulain, A. Monod and H. Wortham, J. Photochem. Photobiol., A, 2007, 187, 10–23 CrossRef CAS PubMed; (b) W. Li, I. Suelves, M. J. Lazaro, S. F. Zhang, T. J. Morgan, A. A. Herod and R. Kandiyoti, Fuel, 2004, 83, 157–179 CrossRef CAS.
- S. A. Hodge, M. K. Bayazit, K. S. Coleman and M. S. P. Shaffer, Chem. Soc. Rev., 2012, 41, 4409–4429 RSC.
- S. Horikoshi, H. Hidaka and N. Serpone, J. Photochem. Photobiol., A, 2001, 138, 69–77 CrossRef CAS.
- A. D. Willey, J. M. Holt, B. A. Larsen, J. L. Blackburn, S. Liddiard, J. Abbott, M. Coffin, R. R. Vanfleet and R. C. Davis, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2014, 32, 5 Search PubMed.
- T. Hasan, V. Scardaci, P. Tan, A. G. Rozhin, W. I. Milne and A. C. Ferrari, J. Phys. Chem. C, 2007, 111, 12594–12602 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, UV-vis spectrum and TGA data. See DOI: 10.1039/c5cc06526g |
| This journal is © The Royal Society of Chemistry 2015 |