
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular tectonics: heterometallic (Ir,Cu) grid-type coordination networks based on cyclometallated Ir(III) chiral metallatectons†
Chaojie
Xu
,
Aurélie
Guenet
*,
Nathalie
Kyritsakas
,
Jean-Marc
Planeix
* and
Mir Wais
Hosseini
*
Molecular Tectonics Laboratory, UMR UDS-CNRS 7140, icFRC, University of Strasbourg, F-67000, Strasbourg, France. E-mail: aguenet@unistra.fr; planeix@unistra.fr; hosseini@unistra.fr
First published on 18th August 2015
Abstract
A chiral-at-metal Ir(III) organometallic metallatecton was synthesised as a racemic mixture and as enantiopure complexes and combined with Cu(II) to afford a heterobimetallic (Ir,Cu) grid-type 2D coordination network.
Molecular chirality in coordination compounds and organometallic complexes1 may be centred on the metal, on the ligand or on both entities. While chiral-at-metal complexes have been employed in homogeneous asymmetric catalysis,2 the use of such complexes in extended architectures such as coordination networks3 has not been fully exploited.4 Coordination networks, also known as coordination polymers or MOFs, are extended periodic architectures resulting from the bridging of consecutives organic entities by metal centres.5 The design of such solid-state crystalline materials may be achieved using the molecular tectonics approach.6 Using self-assembly processes, the generation of coordination networks takes place between programmed organic coordinating tectons and metal centres or complexes behaving as connecting nodes. Homochiral coordination networks based on the use of enantiopure organic tectons have been reported.7,8 Chiral coordination complexes bearing peripheral coordinating sites and thus behaving as metallatectons9 have also been used for the formation of homochiral heterometallic networks.7a–d,10 However, for the reported architectures, the chirality originates from the organic part (mainly BINAP, BINOL or salen-type ligands). Thus, the design and formation of chiral-at-metal coordination networks based on chiral octahedral tris-chelate metallatectons remains a challenge.
Herein, we report the synthesis and characterisation in solution and in the solid state of an unprecedented cyclometallated Ir(III) complex-based metallatecton [Ir(ppy)2(1)][PF6] as a racemic mixture of both Λ and Δ enantiomers, rac-[Ir(ppy)2(1)][PF6], and as enantiopure Λ (Λ-[Ir(ppy)2(1)][PF6]) or Δ (Δ-[Ir(ppy)2(1)][PF6]) complexes (Scheme 1). Furthermore, we describe the formation of a heterometallic (Cu,Ir) 2D grid-type coordination network resulting from the combination of rac-[Ir(ppy)2(1)][PF6] and [Cu(CH3CN)4][BF4].
 | ||
| Scheme 1 Synthetic routes for the preparation of (a) ligand 1 and (b) [Ir(ppy)2(1)][PF6] as a racemate, rac-[Ir(ppy)2(1)][PF6], and enantiomerically pure Δ-[Ir(ppy)2(1)][PF6]. Synthesis of Λ-[Ir(ppy)2(1)][PF6] was performed using D-serine instead of L-serine. The chloride salts of the racemic mixture as well as the enantiopure complexes can be isolated before performing the last metathesis step using KPF6 (step (ii) (2)). | ||
We propose to use chiral-at-metal metallatectons to generate homochiral heterometallic coordination networks. The chiral metallatecton is based on an achiral bipyridyl chelate (bpy) bearing two peripheral monodentate pyridyl coordinating sites. The chirality, of the Λ and Δ type, is introduced through the binding by the chelate moiety of a chiral octahedral metal complex offering two free coordination sites. In order to prevent epimerisation or racemisation, a cyclometallated Ir(III) complex was chosen as configurationally stable entity. Thus, the chiral cationic metallatecton [Ir(ppy)2(1)]+ was designed. The latter combines the organic tecton 1, based on the 2,2′-bipyridine backbone equipped at positions 5 and 5′ with two pyridyl units connected through ethynyl spacers using positions 4 and 4′, with Ir(III) centre complexed by two 2-phenylpyridine (ppy) moieties. The ethynyl spacer was used to extend conjugation of the tecton 1. The choice of a cyclometallated Ir(III) complex was based on its rich photophysical properties.11
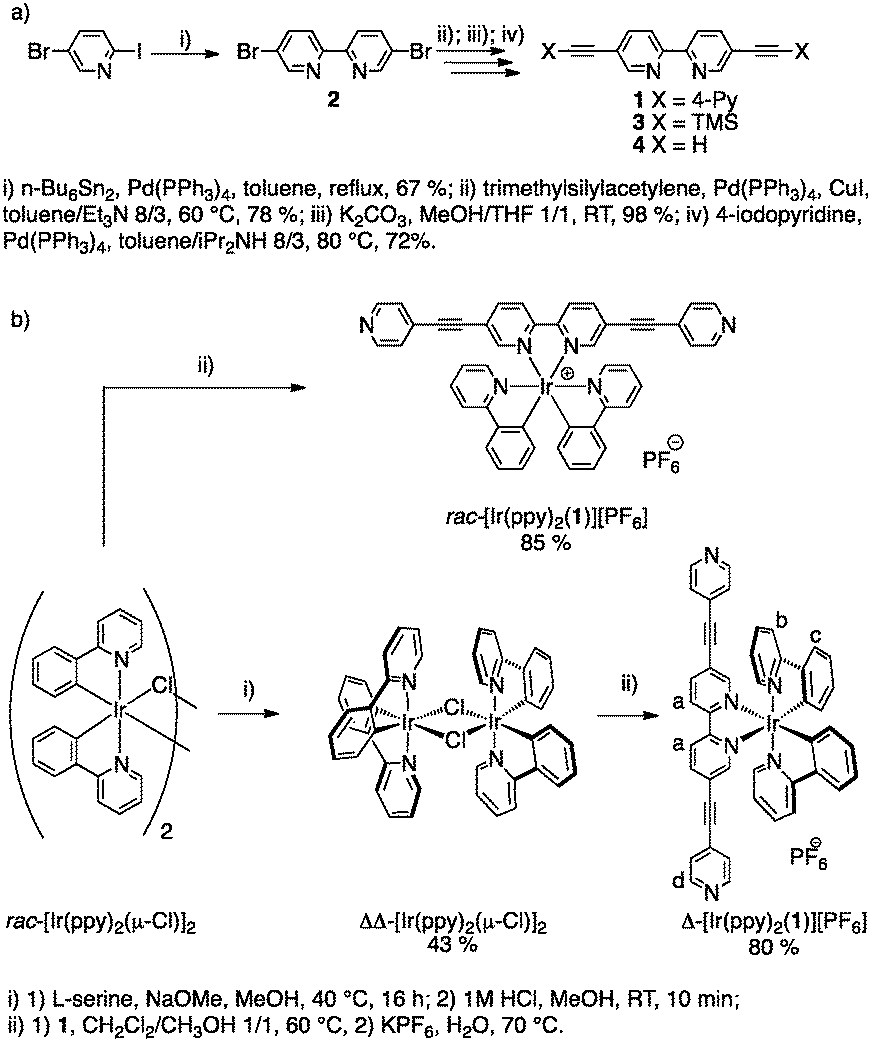
The synthesis of the chiral Ir metallatecton [Ir(ppy)2(1)]+ is based on the precursor tecton 1 (Scheme 1a). The latter was obtained in four steps (37% overall yield) using protection/deprotection and coupling reactions. Starting from 5-bromo-2-iodopyridine, compound 4 was obtained in three steps (51% overall yield).12 A Pd-catalyzed Sonogashira coupling reaction between 4 and 4-iodopyridine afforded the tecton 1 in 72% yield (see ESI†). The Ir(III) complex (rac-[Ir(ppy)2(1)][PF6]), as a mixture of Λ and Δ enantiomers, was prepared in 85% yield (see ESI†) by reaction of the dichloro-bridged Ir(III) dimer rac-[Ir(ppy)2(μ-Cl)]213 with 1 (Scheme 1b).14 For the synthesis of the two enantiopure complexes (Δ- and Λ-[Ir(ppy)2(1)][PF6]), chiral resolution of the Ir(III) dimer was achieved using L or D-serine as chiral auxiliaries (Scheme 1b).15 The two ΔΔ- and ΛΛ-[Ir(ppy)2(μ-Cl)]2 enantiomers were obtained in 43 and 48% yields respectively. The final enantiopure metallatectons Δ- and Λ-[Ir(ppy)2(1)][A] (A = Cl or PF6) were obtained upon reaction of ΔΔ- or ΛΛ-[Ir(ppy)2(μ-Cl)]2 dimer with 1. Both enantiomers were isolated as their chloride salts (74% and 76% for the Δ and the Λ enantiomers respectively). They were converted into their PF6 salts by metathesis with KPF6 in water (80% and 83% yield for the Δ and Λ enantiomers respectively). The two chiral complexes were characterised by standard techniques (see ESI†) and by circular dichroism (CD). As expected, the enantiomers exhibit mirror images CD spectra and opposite specific rotation ([α]D). The racemic mixture as well as enantiopure metallatectons were characterised by X-ray diffraction on single crystals (see ESI†). Single crystals of the racemate rac-[Ir(ppy)2(1)][PF6] were obtained by vapour diffusion of Et2O into a CH3CN solution of rac-[Ir(ppy)2(1)][PF6] (Fig. 1). For the enantiopure chloride salts Δ- and Λ-[Ir(ppy)2(1)][Cl], single crystals could be obtained for both enantiomers upon slow diffusion of toluene into a 1/1 toluene/CH3CN solution of the desired complex (see ESI†) confirming the absolute configuration of the enantiomers. For all three crystals, owing to the severe disorder of solvent molecules, the SQUEEZE command16 was used to remove the corresponding electron density and solve the structure. rac-[Ir(ppy)2(1)][PF6] crystallizes in the triclinic system (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). The geometrical parameters (see ESI†) are similar to those reported for similar iridium(III) complexes containing ppy and bpy ligands.17 The Ir center adopts a slightly distorted octahedral geometry with the expected trans- and cis-arrangement of N and C atoms of the cyclometallated ligand respectively.11b,18 All three chelating ligands are almost planar (average dihedral angle N–C–C–N of ca. −0.322° for the bpy and N–C–C–C angle in the range −6.480° to 1.956° for ppy). The ethynyl spacers are almost linear (C–C–C angles in the 175.36–179.02° range) and the terminal pyridyl units almost coplanar to the bpy unit. As expected, both Δ and Λ enantiomers are present in the crystal. π–π interactions between adjacent complexes are observed in the crystal lattice (see ESI†). Finally, the length of the metallatecton, i.e. the distance between the two terminal N atoms, is ca. 20.6 Å. Both pure Δ- and Λ-[Ir(ppy)2(1)][Cl] enantiomers crystallize in the P21212 chiral space group. The geometrical parameters observed for both enantiomers (see ESI†) are similar to those obtained for the racemic mixture.
). The geometrical parameters (see ESI†) are similar to those reported for similar iridium(III) complexes containing ppy and bpy ligands.17 The Ir center adopts a slightly distorted octahedral geometry with the expected trans- and cis-arrangement of N and C atoms of the cyclometallated ligand respectively.11b,18 All three chelating ligands are almost planar (average dihedral angle N–C–C–N of ca. −0.322° for the bpy and N–C–C–C angle in the range −6.480° to 1.956° for ppy). The ethynyl spacers are almost linear (C–C–C angles in the 175.36–179.02° range) and the terminal pyridyl units almost coplanar to the bpy unit. As expected, both Δ and Λ enantiomers are present in the crystal. π–π interactions between adjacent complexes are observed in the crystal lattice (see ESI†). Finally, the length of the metallatecton, i.e. the distance between the two terminal N atoms, is ca. 20.6 Å. Both pure Δ- and Λ-[Ir(ppy)2(1)][Cl] enantiomers crystallize in the P21212 chiral space group. The geometrical parameters observed for both enantiomers (see ESI†) are similar to those obtained for the racemic mixture.
 | ||
| Fig. 1 Crystal structure of rac-[Ir(ppy)2(1)][PF6]. Solvent molecules, H atoms and PF6 anions have been omitted for clarity. For bond distances and angles see ESI.† | ||
The photophysical properties of the metallatecton as a racemic mixture were studied in the solid state (Fig. 2) and in solution (see ESI†). In solution, the Ir(III) complex exhibits an intense absorption band between 240 and 400 nm attributed to spin-allowed LC transitions involving the ppy and the bpy moieties with a contribution of MLCT and LLCT transitions as previously described for analogous compounds.17a The relatively intense absorption band around 300–360 nm is probably due to π–π* transitions and conjugation over the bpy moieties as already observed on similar complexes.19 In the solid state, a strong absorption band between 250 and 700 nm arising from LC, MLCT and LLCT transitions is observed. The emission spectra recorded for rac-[Ir(ppy)2(1)][PF6] in the solid state (Fig. 2) and in degassed THF (see ESI†) show an unstructured broad band with a maximum at ca. 677 nm (695 nm in degassed THF), characteristic of charge transfer electronic transitions. As for the analogous compound [Ir(ppy)2(bpy)][PF6], the emission may be attributed to a mixture of 3MLCT (Ir → bpy) and 3LLCT (ppy → bpy) transitions.17a,20
 | ||
| Fig. 2 Diffuse reflectance (solid) and emission (dotted) spectra for rac-[Ir(ppy)2(1)][PF6] in the solid state (λexc = 450 nm). | ||
The bathochromic shift observed when compared to [Ir(ppy)2(bpy)][PF6] (λem = 590 nm, RT, THF)19a may be attributed to delocalisation of the bipyridine π system, stabilizing the LUMO as described in solution for ppy-cyclometallated Ir complexes bearing 2,2′-bipyridine ligand substituted in positions 5 and 5′ with aryl moieties17c or oligo(arylene ethynylene) units.19a,c Finally, in the solid state, a quantum yield (QY) of 3% was determined. This relatively weak QY might be related to the presence of π–π interactions, as evidenced in the crystal structure, quenching partially the solid state emission. It should be noted that the PXRD pattern of the sample used for the luminescence measurements matches the simulated pattern from single crystal structural data (see ESI†). Unfortunately, owing to its rather photo-instability in solution, QY value could not be determined accurately.
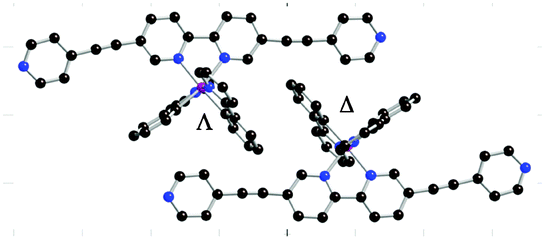
The formation of extended heterometallic coordination networks was explored by combining the Ir(III)-based metallatectons with different metal salts. Only the combination of the racemic mixture rac-[Ir(ppy)2(1)][PF6] and a copper salt afforded single crystals. Indeed, upon slow diffusion of an EtOH solution of [Cu(CH3CN)4][BF4] into a CHCl3 solution of rac-[Ir(ppy)2(1)][PF6], red crystals were obtained after a few days. Structural studies by X-ray diffraction on single crystal revealed that the crystal (space group C2/c) was composed of [Ir(ppy)2(1)]+, Cu2+ cation, BF4− and PF6− anions and solvent molecules (CHCl3, EtOH). Owing to the severe disorder of other solvent molecules, the SQUEEZE command16 was employed to process the data. It appears that Cu(I) was oxidized to Cu(II) during the crystallisation process taking place in the presence of air. The structural investigation revealed the formation of a grid-type network (Fig. 3). The iridium complex within the 2D network exhibits similar geometrical parameters (average bond lengths and angles: Ir–Cppy 2.00 Å, Ir–Nppy 2.04 Å, Ir–Nbpy 2.13 Å, Nppy–Ir–Nppy 174.6°, Nppy–Ir–Cppy 81.0°, Nbpy–Ir–Nbpy 76.5°) as those observed for the discrete metallatecton. However, while in the crystal structure of rac-[Ir(ppy)2(1)][PF6], the terminal pyridyl units were almost coplanar to the backbone, here, they are tilted with respect to the bpy ligand (tilt angles of −77.74° and −31.10°). The Cu(II) centre adopts a slightly distorted octahedral geometry with four pyridyl units belonging to four different metallatectons occupying the square base of the octahedron (Cu–N ca. 2.01 Å) and two BF4− anions in axial positions (Cu–F 2.377(5) Å; F–Cu–F 176.0(2)° and N–Cu–F in the 85.7(2)–91.7(2)° range). Within the 2D-network, two consecutive Cu(II) nodes are separated by 24.44 Å.
 | ||
| Fig. 3 Portions of the crystal structure of rac-Ir(ppy)2(1)·Cu(BF4)2 showing the grid type architecture resulting from the interconnection of the Δ-Ir metallatecton with Cu(BF4)2 acting as metallic nodes (a) and the staggered packing of two consecutive chiral grids along the c axis (b). The chiral grid composed of Λ enantiomers is depicted in red and the one with the Δ enantiomers in blue. Solvent molecules, H atoms and PF6 anions have been omitted for clarity. | ||
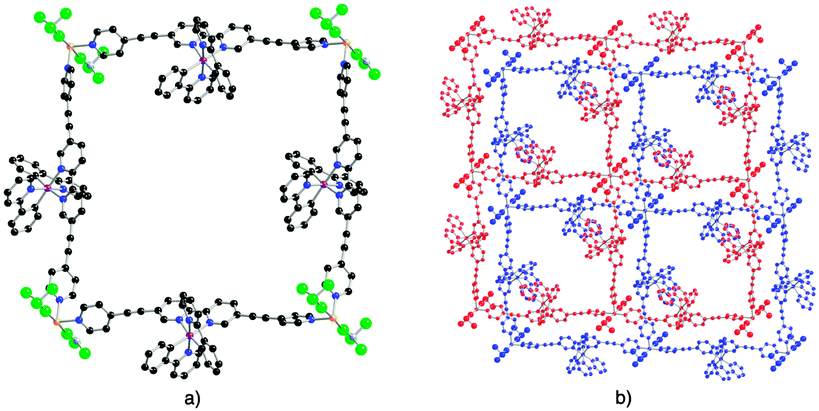
Furthermore, unlike what has been observed before for another grid-like heterometallic coordination network,21 each grid is chiral as only one of the two enantiomers is present (Fig. 3b). The consecutive homochiral sheets are packed in a staggered fashion along the c axis with alternation of Δ and Λ chirality leading thus to a non-chiral crystal. The inter-sheet distance (distance between two mean planes defined by two consecutive grids) is 6.21 Å (see ESI†). Owing to the acentric nature of the metallatecton [Ir(ppy)2(1)]+, within the grid type 2D network, Ir(III) centres may be oriented inwardly (i) or outwardly (o). Thus, for a single grid, four arrangements (i4, i3o, ioio and iioo) are possible (Fig. 4a–d). Furthermore, for the iioo arrangement (Fig. 4d), two types of polar sheets may be formed (Fig. 4e and f). For the heterometallic (Ir,Cu) grid-type architecture discussed above, each sheet is polar (Fig. 4e). However, consecutive sheets are packed in a head-to-tail fashion (centrosymmetric packing) leading thus to an apolar crystal.
 | ||
| Fig. 4 Schematic representations of all four possibilities (a) (i4), (b) (i3o), (c) (ioio) and (d) (iioo) with i and o in and out positioning of the iridium centre within the grid. Green and yellow spheres represent the 4-connecting Cu node and chiral Ir complex respectively. In the case of (d) (iioo), two possibilities of translation lead to polar organisations (e and f). | ||
Unfortunately, crystals were found to be rather air-unstable and thus no proper match between observed and simulated patterns could be obtained by PXRD study (see ESI†). Degradation in less than an hour of the single crystals was also observed using an optical microscope. This might be due to desolvation and collapse of the architecture.
Preliminary photoluminescence measurements (see ESI,† page S3) in the solid state under ambient conditions were carried out on a standard fluorimeter. Unfortunately, no emission of the coordination network could be detected. The luminescence quenching is probably due to the presence of the paramagnetic Cu(II) cation22 or to intermolecular interactions. O2 quenching may also be expected as previously described.23 Because of the air instability of crystals, further studies in view of rationalising the emission quenching could not be carried out.
In conclusion, chiral-at-metal bis-cyclometallated cationic Ir(III) complexes bearing 5,5′-substituted 2,2′-bipyridine as third ligand were synthesised both as a racemic mixture and as enantiomerically pure species and characterised in particular in the solid state by X-ray diffraction on single crystals. The combination of the racemic mixture of Λ and Δ enantiomers behaving as a chiral metallatecton with Cu(II) acting as a 4-connecting node resulted in the formation of an achiral heterobimetallic grid-type coordination network. Unfortunately, the crystalline material appeared to be rather unstable outside the mother liquor. With the aim of generating stable homochiral and luminescent coordination networks, combinations of enantiomerically pure (Λ or Δ) metallatectons [Ir(ppy)2(1)]+ with other metallic salts are currently under investigation.
This contribution is part of an international research project, Chiranet, funded by the European Regional Development Fund (ERDF) under the INTERREG IV Upper Rhine Programme and as part of the Science Offensive of the Trinational Upper Rhine Metropolitan Region. Financial support from the University of Strasbourg, the International Centre for Frontier Research in Chemistry (icFRC), Laboratory of excellence LabEx CSC, Strasbourg, the Institut Universitaire de France, the CNRS is acknowledged.
Notes and references
- (a) A. Werner and V. King, Ber. Dtsch. Chem. Ges., 1911, 44, 1887–1898 CrossRef CAS PubMed; (b) A. von Zelewsky, Stereochemistry of Coordination Compounds, J. Wiley & Sons, Chichester, 1996 Search PubMed; (c) J. Crassous, Chem. Soc. Rev., 2009, 38, 830–845 RSC; (d) E. Meggers, Eur. J. Inorg. Chem., 2011, 2911–2926 CrossRef CAS PubMed; (e) J. Crassous, Chem. Commun., 2012, 48, 9687–9695 RSC; (f) E. C. Constable, Chem. Soc. Rev., 2013, 42, 1637–1651 RSC.
- (a) M. Fontecave, O. Hamelin and S. Ménage, in Topics in Organometallic Chemistry, ed. M. Lemaire and P. Mangeney, Springer, Berlin Heidelberg, 2005, ch. 136353, vol. 15, pp. 271–288 Search PubMed; (b) E. B. Bauer, Chem. Soc. Rev., 2012, 41, 3153–3167 RSC; (c) Z.-Y. Cao, W. D. G. Brittain, J. S. Fossey and F. Zhou, Catal.: Sci. Technol., 2015, 5, 3441–3451 RSC; (d) H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L.-A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100–103 CrossRef CAS PubMed.
- B. F. Abrahams, B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1991, 113, 3606–3607 CrossRef CAS.
- (a) J. Chen and F. M. MacDonnell, Chem. Commun., 1999, 2529–2530 RSC; (b) S. J. Garibay, J. R. Stork, Z. Wang, S. M. Cohen and S. G. Telfer, Chem. Commun., 2007, 4881–4883 RSC.
- (a) Chem. Rev., 2012, 112, MOFs special issue; (b) Chem. Soc. Rev., 2014, 43, themed issue on MOFs.
- (a) M. Simard, D. Su and J. D. Wuest, J. Am. Chem. Soc., 1991, 113, 4696–4698 CrossRef CAS; (b) S. Mann, Nature, 1993, 365, 499–505 CrossRef CAS PubMed; (c) M. W. Hosseini, Acc. Chem. Res., 2005, 38, 313–323 CrossRef CAS PubMed; (d) M. W. Hosseini, CrystEngComm, 2004, 6, 318–322 RSC.
- (a) K. Kim, M. Banerjee, M. Yoon and S. Das, in Topics in Current Chemistry, ed. M. Schröder, Springer, Berlin Heidelberg, 2010, ch. 7, vol. 293, pp. 115–153 Search PubMed; (b) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed; (c) W. Lin, Top. Catal., 2010, 53, 869–875 CrossRef CAS; (d) G. Nickerl, A. Henschel, R. Grünker, K. Gedrich and S. Kaskel, Chem. Ing. Tech., 2011, 83, 90–103 CrossRef CAS PubMed; (e) K. K. Bisht, B. Parmar, Y. Rachuri, A. C. Kathalikattil and E. Suresh, CrystEngComm, 2015, 17, 5341–5356 RSC.
- (a) A. Jouaiti, M. W. Hosseini and N. Kyritsakas, Chem. Commun., 2002, 1898–1899 RSC; (b) P. Grosshans, A. Jouaiti, V. Bulach, J.-M. Planeix, M. W. Hosseini and J.-F. Nicoud, Chem. Commun., 2003, 1336–1337 RSC; (c) A. Jouaiti, M. W. Hosseini, N. Kyritsakas, P. Grosshans and J.-M. Planeix, Chem. Commun., 2006, 3078–3080 RSC; (d) P. Larpent, A. Jouaiti, N. Kyritsakas and M. W. Hosseini, Chem. Commun., 2013, 49, 4468–4470 RSC; (e) N. Marets, V. Bulach and M. W. Hosseini, New J. Chem., 2013, 37, 3549–3558 RSC; (f) P. Larpent, A. Jouaiti, N. Kyritsakas and M. W. Hosseini, Dalton Trans., 2014, 43, 166–172 RSC; (g) P. Larpent, A. Jouaiti, N. Kyritsakas and M. W. Hosseini, Dalton Trans., 2014, 43, 2000–2006 RSC.
- (a) M. W. Hosseini, Chem. Commun., 2005, 5825–5829 RSC; (b) S. Kitagawa, S.-i. Noro and T. Nakamura, Chem. Commun., 2006, 701–707 RSC; (c) M. Andruh, Chem. Commun., 2007, 2565–2577 RSC; (d) S. J. Garibay, J. R. Stork and S. M. Cohen, in Progress in Inorganic Chemistry, John Wiley & Sons, Inc., 2009, ch. 4, vol. 56, pp. 335–378 Search PubMed; (e) S. A. Baudron, CrystEngComm, 2010, 12, 2288–2295 RSC; (f) A. D. Burrows, CrystEngComm, 2011, 13, 3623–3642 RSC; (g) G. Kumar and R. Gupta, Chem. Soc. Rev., 2013, 42, 9403–9453 RSC.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- (a) R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178–8211 CrossRef CAS PubMed; (b) L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, in Topics in Current Chemistry, ed. V. Balzani and S. Campagna, Springer, Berlin Heidelberg, 2007, ch. 131, vol. 281, pp. 143–203 Search PubMed.
- J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15–20 CrossRef CAS PubMed.
- S. Sprouse, K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1984, 106, 6647–6653 CrossRef CAS.
- Adapted from: L. L. Tinker, N. D. McDaniel, E. D. Cline and S. Bernhard, Inorg. Synth., 2010, 35, 168–173 CAS.
- O. Chepelin, J. Ujma, X. Wu, A. M. Z. Slawin, M. B. Pitak, S. J. Coles, J. Michel, A. C. Jones, P. E. Barran and P. J. Lusby, J. Am. Chem. Soc., 2012, 134, 19334–19337 CrossRef CAS PubMed.
- A. L. Spek, PLATON, The University of Utrecht, Utrecht, The Netherlands, 1999 Search PubMed.
- For example see (a) R. D. Costa, E. Ortí, H. J. Bolink, S. Graber, S. Schaffner, M. Neuburger, C. E. Housecroft and E. C. Constable, Adv. Funct. Mater., 2009, 19, 3456–3463 CrossRef CAS PubMed; (b) T. Moriuchi, C. Katano and T. Hirao, Chem. Lett., 2012, 41, 310–312 CrossRef CAS; (c) S. Ladouceur, D. Fortin and E. Zysman-Colman, Inorg. Chem., 2010, 49, 5625–5641 CrossRef CAS PubMed; (d) F. Neve, M. La Deda, A. Crispini, A. Bellusci, F. Puntoriero and S. Campagna, Organometallics, 2004, 23, 5856–5863 CrossRef CAS.
- F. O. Garces, K. A. King and R. J. Watts, Inorg. Chem., 1988, 27, 3464–3471 CrossRef CAS.
- (a) K. D. Glusac, S. Jiang and K. S. Schanze, Chem. Commun., 2002, 2504–2505 RSC; (b) F. Lafolet, S. Welter, Z. Popovic and L. D. Cola, J. Mater. Chem., 2005, 15, 2820–2828 RSC; (c) K.-Y. Kim, R. T. Farley and K. S. Schanze, J. Phys. Chem. B, 2006, 110, 17302–17304 CrossRef CAS PubMed.
- (a) M. G. Colombo, A. Hauser and H. U. Guedel, Inorg. Chem., 1993, 32, 3088–3092 CrossRef CAS; (b) Y. Ohsawa, S. Sprouse, K. A. King, M. K. DeArmond, K. W. Hanck and R. J. Watts, J. Phys. Chem., 1987, 91, 1047–1054 CrossRef CAS.
- (a) A. Béziau, S. A. Baudron, D. Rasoloarison and M. W. Hosseini, CrystEngComm, 2014, 16, 4973–4980 RSC; (b) A. Béziau, S. A. Baudron, G. Rogez and M. W. Hosseini, Inorg. Chem., 2015, 54, 2032–2039 CrossRef PubMed.
- (a) K. Jayaramulu, R. P. Narayanan, S. J. George and T. K. Maji, Inorg. Chem., 2012, 51, 10089–10091 CrossRef CAS PubMed; (b) P. Wu, J. Wang, C. He, X. Zhang, Y. Wang, T. Liu and C. Duan, Adv. Funct. Mater., 2012, 22, 1698–1703 CrossRef CAS PubMed.
- (a) Y.-T. Chen, C.-Y. Lin, G.-H. Lee and M.-L. Ho, CrystEngComm, 2015, 17, 2129–2140 RSC; (b) M.-L. Ho, Y.-A. Chen, T.-C. Chen, P.-J. Chang, Y.-P. Yu, K.-Y. Cheng, C.-H. Shih, G.-H. Lee and H.-S. Sheu, Dalton Trans., 2012, 41, 2592–2600 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, analytical (NMR, CD, UV, emission spectra) and crystallographic data. CCDC 1413248–1413251. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc06427a |
| This journal is © The Royal Society of Chemistry 2015 |