
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkynol natural products target ALDH2 in cancer cells by irreversible binding to the active site†
Wolfgang
Heydenreuter
,
Elena
Kunold
and
Stephan A.
Sieber
*
Department of Chemistry, Center for Integrated Protein Science Munich (CIPSM), Technische Universität München, Garching, Germany. E-mail: stephan.sieber@tum.de
First published on 7th September 2015
Abstract
Falcarinol and stipudiol are natural products with potent anti-cancer activity found in several vegetables. Here, we use a chemical proteomic strategy to identify ALDH2 as a molecular target of falcarinol in cancer cells and confirm enzyme inhibition via covalent alkylation of the active site. Furthermore, the synthesis of stipudiol led to the observation that ALDH2 exhibits preference for alkynol-based binders. Inhibition of ALDH2 impairs detoxification of reactive aldehydes and limits oxidative stress response, two crucial pathways for cellular viability.
In addition to their obvious nutritional value, vegetables represent a largely uncharacterized source of bioactive natural products. Of these, alkynols represent a prominent compound class featuring long hydrophobic chains with consecutive triple bonds and a stereogenic hydroxyl group.1 Among the most biologically active members of this class are (+/−)-falcarinol (FL1) and stipudiol (ST) (Fig. 1).2,3 Falcarinol, a component of carrots, parsley and ginseng, exhibits potent anti-cancer activities that have been linked to vegetable- associated health benefits.4 However, falcarinol has also been described as a contact allergen, suggesting that it is capable of irreversibly attaching to proteins.5,6 While the biological importance of falcarinol has been subject to several studies, no detailed mechanistic analysis of the molecular mode of action has been performed until now. Falcarinol exhibits limited stability in aqueous solution and is prone to a multitude of possible alkylation reactions with nucleophiles.6,7 Studies of falcarinol with isolated proteins have revealed that the compound alkylates cannabinoid receptor 16 and modulates the activity of ABCG2 (ATP-binding cassette sub-family G member 2)8 as well as GABAA receptors;7 however, it is unknown whether these interactions also occur in a cellular context. Kaiser et al. recently studied the molecular targets of callyspongynic acid, a different polyacetylene natural product, in human cells. A suite of metabolic enzymes was identified that are involved in the metabolism and degradation of lipids and fatty acids.9 Here, we prepare the first falcarinol probes capable of bioorthogonal ligation to affinity tags for proteome-wide target identification via quantitative mass spectrometry.10–12 Our studies confirm ALDH2 as a specific target that irreversibly binds to the electrophilic core scaffold of falcarinol via a nucleophilic cysteine. Furthermore, we describe the first total synthesis of stipudiol, a related natural product, and confirm a conserved preference of alkynol-based natural products for ALDH2 inhibition.
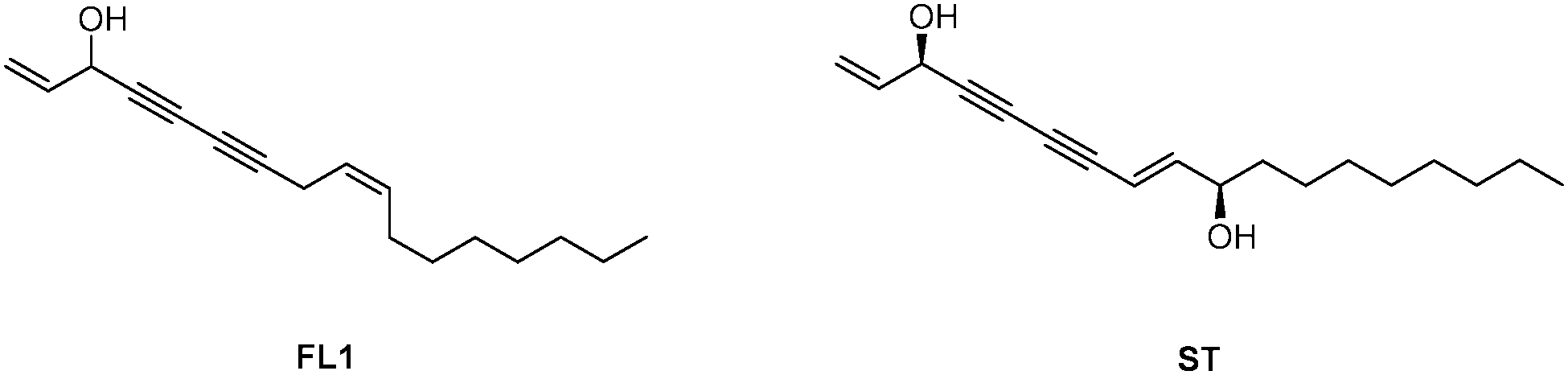 | ||
| Fig. 1 (+/−)-Falcarinol (FL1) and stipudiol (ST). | ||
To investigate cellular falcarinol targets, we utilized an established synthesis to generate the natural product (FL1),7 its corresponding alkyne-modified probe (FL2) and an oxidized probe mimicking the natural product falcarinone (FL3) (Scheme 1). 1,8-Octanediol was converted to diyne 2via iodination und subsequent alkynylation. FL2 was obtained as the coupling product of 5-bromo-pent-1-en-4-yn-3-ol (3) with 2 in a Chadiot–Chodkiewicz reaction. Allylic oxidation with activated MnO2 yielded FL3. Importantly, FL1, FL2 and FL3 exhibited comparable cytotoxic activity against HepG2 and A549 cells with IC50 values of 1–3 μM, suggesting that minor modifications do not significantly alter biological activity (Table S1, ESI†). Furthermore, the obtained IC50 values are in accordance with previous cytotoxicity studies.1,13,14
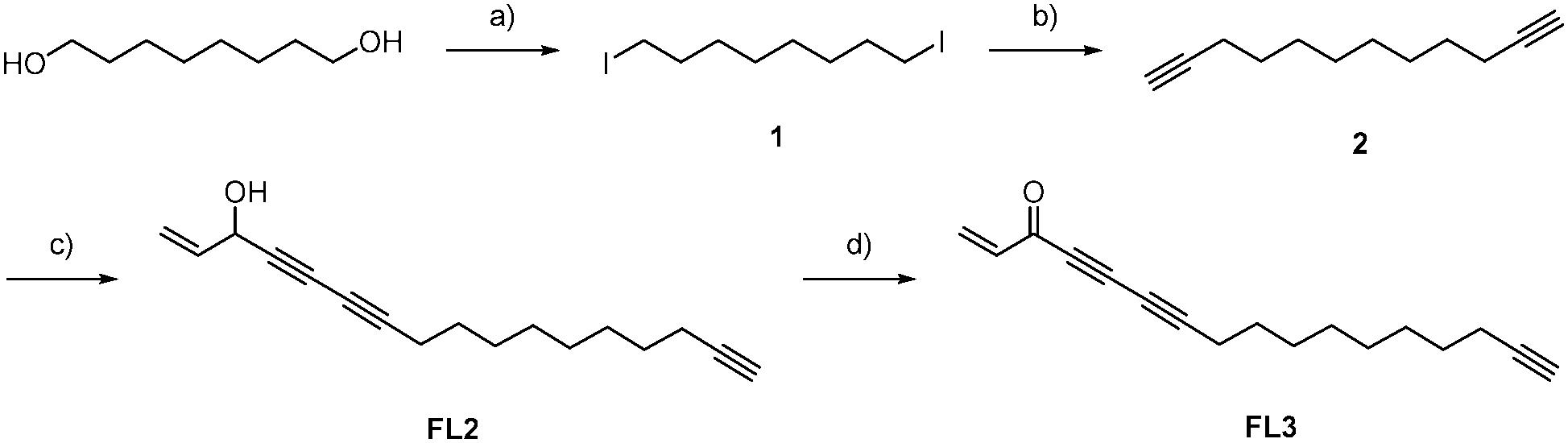 | ||
| Scheme 1 Synthesis of FL2 and FL3: (a) 1. NEt3 (4.00 eq.), TsCl (2.10 eq.), CH2Cl2, 2. NaI (2.00 eq.), acetone, 0 °C → rt, 16 h, 71%; (b) 1. ethynyltrimethyl-silane (2.30 eq.), nBuLi (2.30 eq.), THF, −78 °C, 2. 1 (1.00 eq.), DMPU, 3. K2CO3 (2.00 eq.), MeOH, rt; (c) NH2OH·HCl, EtNH2 (70%), CuCl (10 mol%), 5-bromopent-1-en-4-yn-3-ol (3, 0.75 eq.), MeOH, CH2Cl2, 0 °C → rt, 4h, 56%; (d) MnO2 (20.0 eq.), Et2O, rt, 4 h, 36%. | ||
For target identification, FL2 was incubated with intact A549 cells for one hour at concentrations similar to the IC50 value (1 and 5 μM). Subsequent cell lysis and bioorthogonal ligation to rhodamine azide via click chemistry,15–17 followed by SDS-PAGE and fluorescent scanning, revealed one prominent enzyme band in the cytosolic fraction at approximately 55–60 kDa (Fig. 2). Interestingly, a similar profile was observed with falcarinone probe FL3, suggesting that both molecules may share a common mechanism of active site modification. This mechanism is likely based on the highly reactive diyne moiety, which is present in both compounds and has been observed to react with thiol nucleophiles at elevated pH.7 At physiological pH no reaction of FL2 with a cysteine thiol was observed (16 h) suggesting that activated thiols (e.g. in active sites) are required for binding (Fig. S1, ESI†). We used two strategies in order to identify both reversible and irreversible cellular targets of falcarinol. First, the probe-treated proteome of A549 cancer cells was clicked to a trifunctional rhodamine–biotin-azide linker18 and enriched using avidin beads. Upon preparative SDS-PAGE (Fig. S2, ESI†), the 60 kDa band was isolated, digested and investigated via mass spectrometry (MS). MS analysis revealed aldehyde dehydrogenase 2 (ALDH2) as a pronounced irreversible target candidate (Fig. S3 and Table S2, ESI†). In our second approach, we designed and synthesized a photoprobe FL2P (Scheme 2), with a photolabile diazirine moiety incorporated next to the alkyne handle, in order to enable the identification of reversible targets.19 The symmetric alcohol 4 was installed by lithiation of (5-iodopent-1-yn-1-yl)trimethylsilane and subsequent addition to the corresponding C-6 aldehyde. After deprotection, oxidation and diazirination, 7 was coupled to 5-bromo-pent-1-en-4-yn-3-ol (3) applying the same conditions as for FL2.
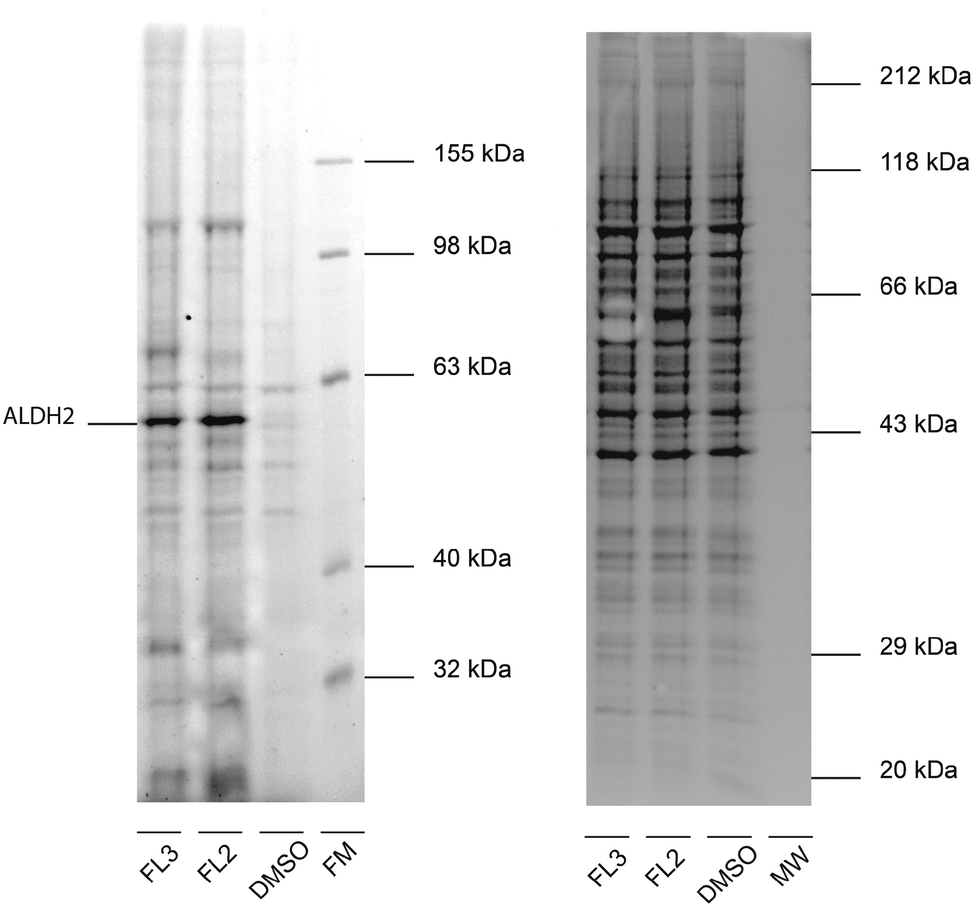 | ||
| Fig. 2 In situ labeling of H. sapiens A549 cells with alkyne-tagged probes. Fluorescent gel (left) and corresponding coomassie stain (right) after labeling with FL2 and FL3 (5 μM, 1 h incubation) and click reaction with rhodamine azide show a prominent band, representing ALDH2 as confirmed via mass spectrometry (FM = fluorescent molecular weight marker, MW = different molecular weight marker for coomassie staining). | ||
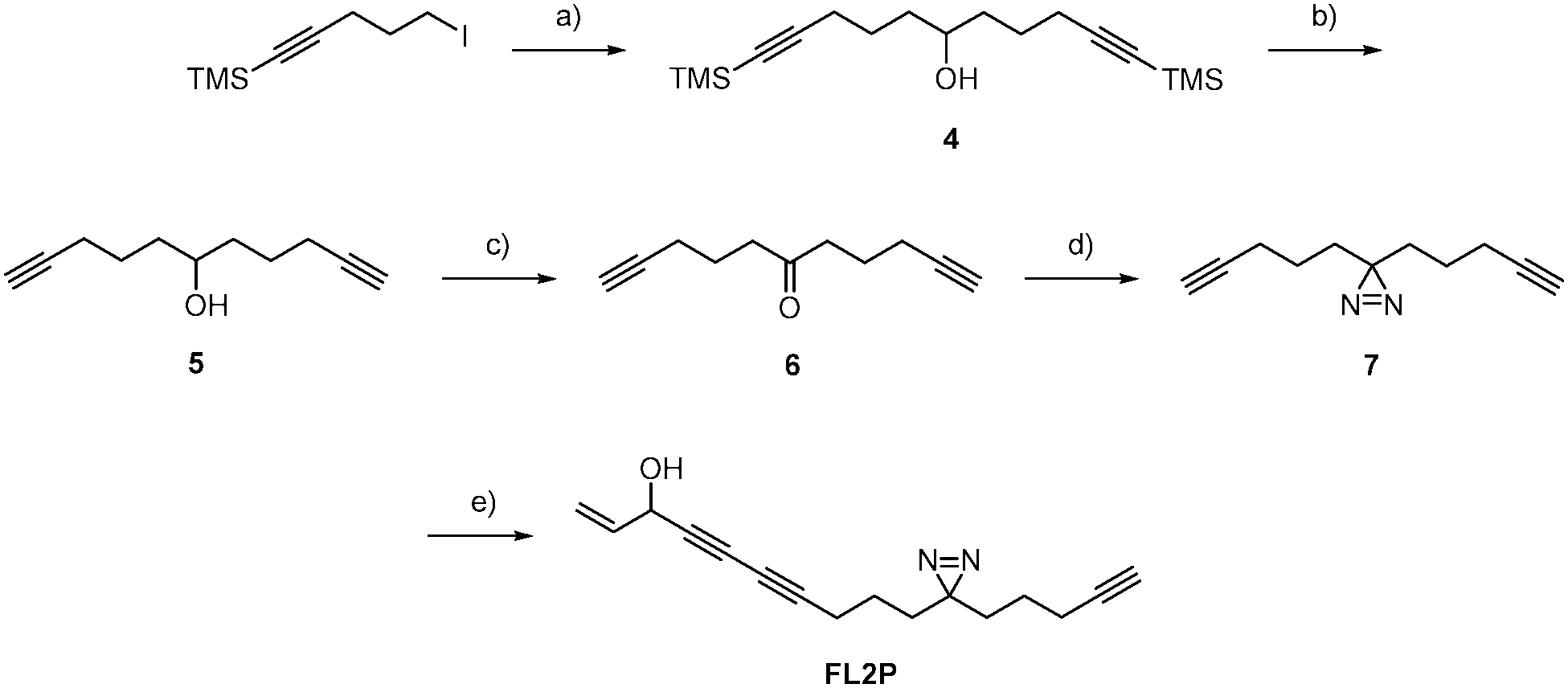 | ||
| Scheme 2 Synthesis of FL2P (a) tBuLi (2.05 eq.), 6-(trimethylsilyl)hex-5-ynal (1.00 eq.), heptane/THF, −78 °C → rt, 16 h, 66%; (b) K2CO3 (2.30 eq.), MeOH, rt, 16 h, quant.; (c) PCC (1.50 eq.), CH2Cl2, rt, 12 h, 83%; (d) 1. NH3 (MeOH) (20.0 eq.), H2NOSO2H (1.30 eq.), 2. I2, MeOH, −10 °C → rt, 16 h, 30%; (e) NH2OH·HCl (0.33 eq.), EtNH2 (70%), CuCl (10 mol%), 5-bromo-pent-1-en-4-yn-3-ol (3, 0.75 eq.), MeOH, CH2Cl2, 0 °C → rt, 4 h, 31%. | ||
To gain a global overview of all covalent and non-covalent FL2P targets in living cells, we utilized a quantitative, gel-free proteomic analysis. Therefore, A549 cells were cultivated in media containing either heavy or light isotope-labeled amino acids (SILAC)20 and incubated with probe or DMSO. Upon UV-irradiation, the cells were lysed and clicked to biotin-azide in order to facilitate subsequent enrichment using avidin beads. The enriched proteins were digested using trypsin and peptides were analyzed via gel-free MS. Again, ALDH2 was among the top hits with an average enrichment ratio of about 4 (Fig. 3 and Table S3, ESI†). In addition, the SILAC studies revealed heme oxygenase 2, homo sapiens translocase of outer mitochondrial membrane 22 homolog (MST065), cytochrome b5 Type B, serum paraoxonase/lactonase 3 (PON3) and cathepsin D (HEL-S-130P) as putative target proteins.
 | ||
| Fig. 3 Quantitative proteome enrichment analysis of FL2P treated A549 cells compared to DMSO treated control after click reaction with biotin azide and pull-down with avidin beads. The volcano plot shows the statistical significance of enrichment levels (student's t-test p-value) as a function of average protein ratios from three biological replicates in probe-treated vs. control cells. Red dots represent proteins with ratios and p-values above 3(log2 corresponds to 1.6) and 0.05(−log10 corresponds to 1.3), respectively. | ||
Due to its pronounced enrichment in gel based and gel-free MS experiments, we validated ALDH2 as a putative falcarinol target by spiking recombinant, purified ALDH2 into A549 cell lysate followed by incubation with FL2. A strong concentration-dependent labeling was observed, which disappeared upon heat-denaturation prior to compound addition and therefore suggests that an active, folded enzyme is required for binding (Fig. S4, ESI†). Intact protein MS revealed that one molecule of FL1 attaches to ALDH2 (Fig. S5, ESI†) and corresponding sequencing via LC-MS/MS showed that a peptide containing Cys318, 319 and 320 is modified with the compound (Fig. S6, ESI†). Although the experiment was repeated several times under different conditions the fragmentation spectra did not allow for an unequivocal assignment of any of the three residues. Interestingly, Cys318 and 320 are located next to the catalytic center of ALDH2 (Cys319) that is crucial for enzyme activity.21 Cys319 helps ALDH2 catalyze the oxidation of aldehydes to carboxylic acids by nucleophilically attacking aldehyde substrates. Thus, either binding to Cys318 or 320 sterically blocks the active site and prevents catalysis or binding to Cys319 directly inhibits activity. Corresponding to this mechanism, inhibition of ALDH2 with 5 μM FL1 and FL2 at rt for 3h fully abolished enzymatic activity in a NAD+-coupled assay (Fig. S7, ESI†). Due to the covalent and irreversible nature of the inhibition, binding kinetics were determined according to Kitz and Wilson via time-dependent inhibition measurements.22 A KI value of 2.96 μM and a kinact value of 0.32 h−1 for FL1 demonstrated potent inhibition comparable to previously described ALDH inhibitors (Fig. 4A).23
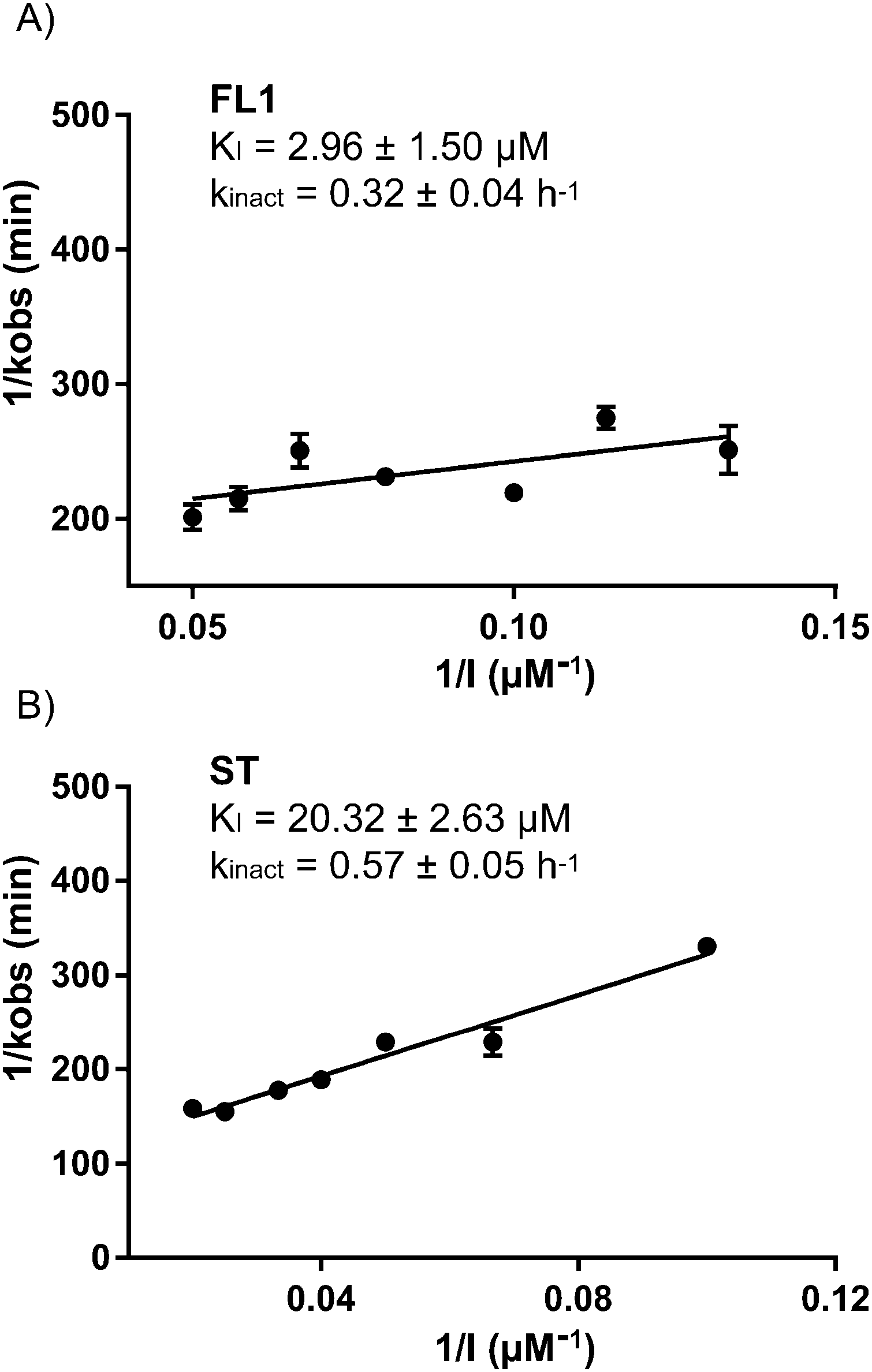 | ||
| Fig. 4 Determination of KI and kinact according to the method of Kitz and Wilson for FL1 (A) and stipudiol (ST) (B) with ALDH2 (n = 3). Recombinant ALDH2 was incubated for varying periods of time and concentrations and the residual activity determined in a coupled assay. The increase of NADH resulting from the metabolism of propionaldehyde was measured at 340 nm. | ||
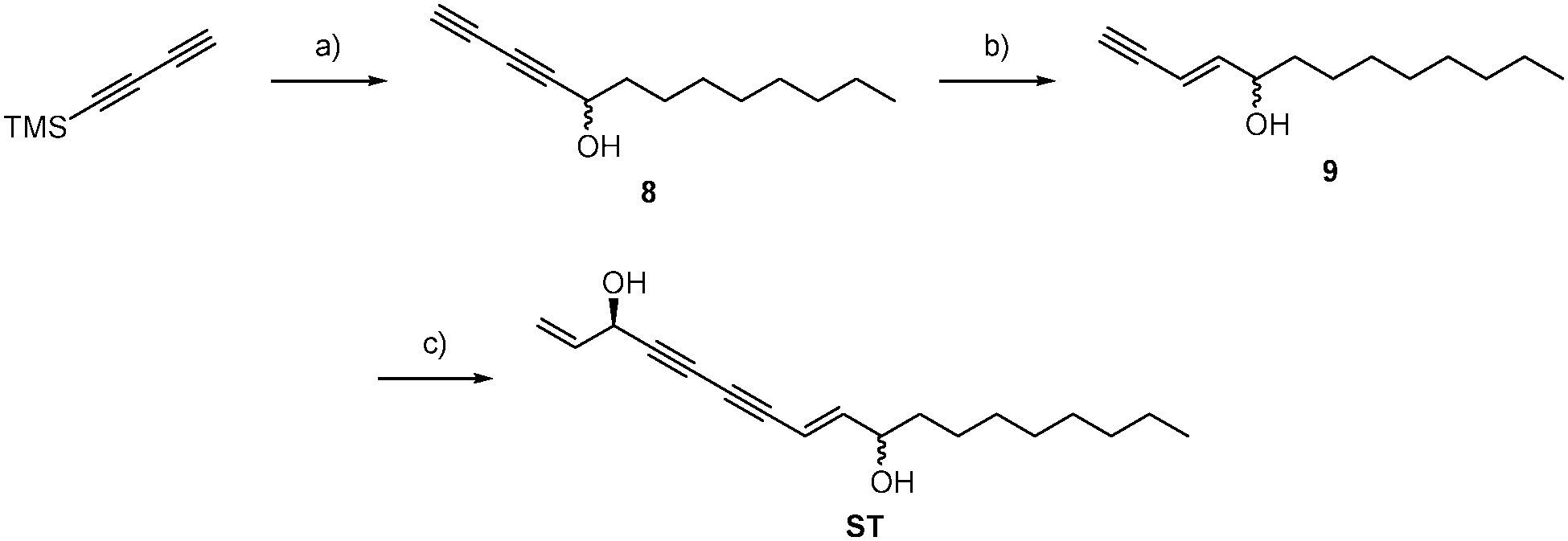
Since ALDH2 exhibits a narrow entry channel that is hypothesized to accommodate long-chain aliphatic compounds (Fig. S8, ESI†), we were interested if related alkynol natural products such as stipudiol (ST) would bind and inhibit ALDH2 as well. We therefore designed a synthetic route to stipudiol that relied on the coupling of two fragments at the diyne moiety using Chadiot–Chodkiewicz conditions. First, we generated the right fragment of stipudiol, starting with the enantioselective addition of TMS-diyne to nonanal24 to generate alkynol 8, which was subsequently reduced to enyne 9 using LiAlH4. For the coupling reaction, optically pure (R)-5-bromopent-1-en-4-yn-3-ol (10) was obtained from 5-(trimethylsilyl)-pent-1-en-4-yn-3-ol according to literature procedure involving chiral resolution with PS Lipase.25 The fragments were coupled as mentioned before for probes FL2 and FL2P (Scheme 3).26ST modified ALDH2 (Fig. S5, ESI†) and inhibited its activity with a KI of 20.32 μM and a kinact of 0.57 h−1 (Fig. 4B). Although the potency of inhibition is reduced compared to falcarinol, our studies prove that ALDH2 represents a mutual target for this natural product class.
 | ||
| Scheme 3 (a) 1. MeLi (1.10 eq.), H2O; 2. (S)-binol (40 mol%), Cy2NH (5 mol%), ZnEt2 (3.00 eq.), 3. Ti(OiPr)4 (1.00 eq.) nonanal (1.00 eq.), 4. 1 M NaOH, Et2O, THF, rt, 20 h, 58%; (b) LiAlH4 (1.20 eq.), THF, 0 °C → rt, 3 h (69%); (c) NH2OH·HCl (1.70 eq.) ethylamine, (R)-5-bromopent-1-en-4-yn-3-ol (10, 1.20 eq.), CuCl (9 mol%), MeOH, 1.5 h, 66%. | ||
Aldehyde dehydrogenases are a broad superfamily of enzymes that is important for the metabolism and detoxification of aldehydes in the body. ALDH2 is not only an essential enzyme for ethanol detoxification but also plays an important role in the metabolism of toxic lipid aldehydes such as 4-hydroxy-2-nonenal.27 Consequently, reduction of ALDH2 activity increases susceptibility to reactive aldehydes and oxidative stress, which cause cell damage and apoptosis.28,29 Thus, the structural similarity between native substrates and falcarinol or stipudiol provides a rational for their inhibition of ALDH2 and likely explains their corresponding cellular toxicity. Although we cannot exclude that additional enzymatic pathways are affected due to inherent detection limits, our results highlight the biological effects of falcarinol and stipudiol and reveal insight into their reactivity within the human body. In addition, we present falcarinol-based probe molecules which can be used as selective tools to manipulate and further study ALDH2, an enzyme with functions beyond alcohol detoxification which require further elucidation.
S.A.S. acknowledges funding by the Deutsche Forschungs-gemeinschaft, FOR1406, the excellence cluster CIPSM. E.K. was supported by Fonds der chemischen Industrie. We would like to thank M. M. Maturi for HPLC analysis of chiral compounds and Annabelle Hoegl for critical proofreading of the manuscript.
Notes and references
- K. Brandt, L. P. Christensen, J. Hansen-Møller, S. L. Hansen, J. Haraldsdottir, L. Jespersen, S. Purup, A. Kharazmi, V. Barkholt, H. Frøkiær and M. Kobæk-Larsen, Trends Food Sci. Technol., 2004, 15, 384–393 CrossRef CAS PubMed.
- L. P. Christensen, Recent Pat. Food, Nutr. Agric., 2011, 3, 64–77 CrossRef CAS.
- C. Liang, Y. Ding, J. A. Kim, S. Y. Yang, H.-J. Boo, H.-K. Kang, M. C. Nguyen and Y. H. Kim, Bull. Korean Chem. Soc., 2011, 32, 3513–3516 CrossRef CAS.
- L. P. Christensen and K. Brandt, J. Pharm. Biomed. Anal., 2006, 41, 683–693 CrossRef CAS PubMed.
- L. Hansen, O. Hammershoy and P. M. Boll, Contact Dermatitis, 1986, 14, 91–93 CrossRef CAS PubMed.
- M. Leonti, L. Casu, S. Raduner, F. Cottiglia, C. Floris, K. H. Altmann and J. Gertsch, Biochem. Pharmacol., 2010, 79, 1815–1826 CrossRef CAS PubMed.
- M. M. Czyzewska, L. Chrobok, A. Kania, M. Jatczak, F. Pollastro, G. Appendino and J. W. Mozrzymas, J. Nat. Prod., 2014, 77, 2671–2677 CrossRef CAS PubMed.
- K. W. Tan, D. P. Killeen, Y. Li, J. W. Paxton, N. P. Birch and A. Scheepens, Eur. J. Pharmacol., 2014, 723, 346–352 CrossRef CAS PubMed.
- S. Nickel, R. A. Serwa, F. Kaschani, S. Ninck, S. Zweerink, E. W. Tate and M. Kaiser, Chem. – Eur. J., 2015, 21, 10721–10728 CrossRef CAS PubMed.
- M. J. Evans and B. F. Cravatt, Chem. Rev., 2006, 106, 3279–3301 CrossRef CAS PubMed.
- M. Fonovic and M. Bogyo, Expert Rev. Proteomics, 2008, 5, 721–730 CrossRef CAS PubMed.
- W. P. Heal, T. H. Dang and E. W. Tate, Chem. Soc. Rev., 2011, 40, 246–257 RSC.
- J. F. Young, S. J. Duthie, L. Milne, L. P. Christensen, G. G. Duthie and C. S. Bestwick, J. Agric. Food Chem., 2007, 55, 618–623 CrossRef CAS PubMed.
- C. Zidorn, K. Johrer, M. Ganzera, B. Schubert, E. M. Sigmund, J. Mader, R. Greil, E. P. Ellmerer and H. Stuppner, J. Agric. Food Chem., 2005, 53, 2518–2523 CrossRef CAS PubMed.
- V. V. Rostovtsev, J. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- R. Huisgen, Proc. Chem. Soc., 1961, 357–396 Search PubMed.
- J. Eirich, J. L. Burkhart, A. Ullrich, G. C. Rudolf, A. Vollmar, S. Zahler, U. Kazmaier and S. A. Sieber, Mol. BioSyst., 2012, 8, 2067–2075 RSC.
- Z. Li, P. Hao, L. Li, C. Y. Tan, X. Cheng, G. Y. Chen, S. K. Sze, H. M. Shen and S. Q. Yao, Angew. Chem., Int. Ed. Engl., 2013, 52, 8551–8556 CrossRef CAS PubMed.
- S. E. Ong, B. Blagoev, I. Kratchmarova, D. B. Kristensen, H. Steen, A. Pandey and M. Mann, Mol. Cell. Proteomics, 2002, 1, 376–386 CAS.
- J. Farres, T. T. Wang, S. J. Cunningham and H. Weiner, Biochemistry, 1995, 34, 2592–2598 CrossRef CAS.
- R. Kitz and I. B. Wilson, J. Biol. Chem., 1962, 237, 3245–3249 CAS.
- T. Wirth, K. Schmuck, L. F. Tietze and S. A. Sieber, Angew. Chem., Int. Ed. Engl., 2012, 51, 2874–2877 CrossRef CAS PubMed.
- M. Turlington, Y. H. Du, S. G. Ostrum, V. Santosh, K. Wren, T. Lin, M. Sabat and L. Pu, J. Am. Chem. Soc., 2011, 133, 11780–11794 CrossRef CAS PubMed.
- T. J. Mann, A. W. H. Speed, R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed. Engl., 2013, 52, 8395–8400 CrossRef CAS PubMed.
- H. D. Yun, T. C. Chou, H. J. Dong, Y. Tian, Y. M. Li and S. J. Danishefsky, J. Org. Chem., 2005, 70, 10375–10380 CrossRef CAS PubMed.
- D. P. Hartley, J. A. Ruth and D. R. Petersen, Arch. Biochem. Biophys., 1995, 316, 197–205 CrossRef CAS PubMed.
- A. D. Ebert, K. Kodo, P. Liang, H. Wu, B. C. Huber, J. Riegler, J. Churko, J. Lee, P. de Almeida, F. Lan, S. Diecke, P. W. Burridge, J. D. Gold, D. Mochly-Rosen and J. C. Wu, Sci. Transl. Med., 2014, 6, 255ra130 CrossRef PubMed.
- K. H. Moon, B. J. Kim and B. J. Song, FEBS Lett., 2005, 579, 6115–6120 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc06424d |
| This journal is © The Royal Society of Chemistry 2015 |