
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Measuring couplings in crowded NMR spectra: pure shift NMR with multiplet analysis†
M.
Foroozandeh
*,
R. W.
Adams
 ,
P.
Kiraly
,
M.
Nilsson
and
G. A.
Morris
,
P.
Kiraly
,
M.
Nilsson
and
G. A.
Morris
School of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK. E-mail: Mohammadali.foroozandeh@manchester.ac.uk
First published on 27th August 2015
Abstract
The PSYCHE method for pure shift NMR is exploited to generate 2D J spectra with full decoupling in one dimension and multiplet structure in the other, allowing spin–spin coupling constants to be measured even in very crowded spectra. Significant improvements over existing techniques are demonstrated for the hormones estradiol and androstenedione.
Resolution and sensitivity are longstanding challenges in 1H NMR spectroscopy. A major limitation on resolution in 1H NMR spectra is the presence of homonuclear scalar coupling between protons, which splits signals into multiplets. Because of the narrow range of proton chemical shifts, multiplet overlap is very common and can severely complicate the analysis and assignment of spectra. In particular, spectral overlap often prevents the measurement of the spin–spin coupling constants that provide essential information on molecular structure and conformation. In the last decade, “pure shift” NMR, in which the multiplet structure of signals due to homonuclear coupling is collapsed to singlets, greatly improving resolution, has become a practical tool. Here an experimental method is described that combines the high chemical shift resolution of pure shift NMR with the measurement of multiplet structure.
Pure shift NMR experiments generally (though not universally) use a pulse sequence element that selectively refocuses the effects of evolution under couplings.1 The elegant method of Zangger and Sterk (ZS),2 which uses a spatially- and frequency-selective 180° pulse, and Pines's much older BIRD3 (bilinear rotation decoupling) method, which exploits one-bond couplings with sparse heteronuclei to refocus all but geminal couplings, are currently the two J-refocusing elements most commonly used in pure shift NMR. These approaches have found applications in 1D NMR,4 DOSY,5 and 2D experiments including TOCSY,4e,6 NOESY,7 2D J spectroscopy,8 and HSQC.9 Unfortunately, the ZS method incurs a very large sensitivity penalty when the chemical shift difference between resonances to be decoupled is small, since then the signals required originate from a correspondingly small fraction of the sample. BIRD has mostly been used with protons directly bonded to 13C at natural abundance, which usually gives a minimum sensitivity penalty of two orders of magnitude. (If, however, BIRD pure shift acquisition is used in an experiment that already relies on dilute heteronuclei, for example in 1H–13C HSQC at natural abundance9a or 1H–15N HSQC in proteins,10 the sensitivity price has already been paid and the method is highly effective).
More recently, a new pure shift pulse sequence element, Pure Shift Yielded by Chirp Excitation (PSYCHE), has been introduced that relies on statistical separation of spin populations by the use of low flip angle (β) swept-frequency chirp pulses. PSYCHE offers almost an order of magnitude improvement in performance over existing methods, as demonstrated for 1D 1H NMR11 and 2D 1H–1H TOCSY12 experiments. Currently, band-selective homodecoupling (BASHD) methods13 are the only competitors for PSYCHE in terms of sensitivity, but these are not broadband, only decoupling part of a spectrum.
Pure shift NMR can greatly facilitate the assignment of crowded spectra. However, it accomplishes this by hiding14 or eliminating12 homonuclear multiplet structure, sacrificing potentially valuable information on structure and bonding. Multiplet structure is often simply a nuisance in crowded 1D spectra, as it cannot be disentangled and reduces, rather than increases, the structural information available. Where multiplet structure can be recovered reliably, however, the coupling constants it makes available can be very useful. The ideal case would be an experiment that produces a pure shift spectrum, with a singlet for each chemical site, but makes high resolution multiplet structure available in a second dimension.
In principle, one such experiment is 2D J (J-resolved) spectroscopy,15 which shows the normal coupled spectrum in F2 but coupling structure only in F1. Unfortunately the phase-twist lineshape in such experiments severely complicates matters, to the extent that it is normal to sacrifice much of the potential resolution advantage of the technique by using severe time-domain weighting functions and absolute value mode display. Because the projection of a phase-twist lineshape at 45° in frequency space is zero, pure shift spectra generated by conventional 2D J spectroscopy likewise require absolute value mode calculation and hence have degraded resolution and severely distorted intensities.
There have been a number of attempts to produce pure shift NMR spectra and extract absorption mode multiplet structure from 2D J spectra, variously using combined data manipulation and pattern recognition,16 modified pulse sequences,8,16,17 and purely data post-processing approaches.18 So far these have not reached practical application, and questions as to their sensitivity, reliability, and suitability for complex spin systems have remained either unanswered or without satisfactory responses. There is thus a clear need for a practical 2D J method that can give clean reliable spectra, with good sensitivity. The great strength of such methods is that they approach the ideal limit of resolution of a 1H NMR spectrum, since multiplet structure in F2 is suppressed and linewidths in F1 are very close to the limiting natural linewidths, B0 inhomogenity contributions being refocused.
The most successful method to date is that of Pell and Keeler.8 They showed that the required pure absorption mode 2D J spectrum can be obtained by using the ZS J-refocusing pulse element to reverse the sense of t1 evolution in a 2D J pulse sequence. Combining normal and reversed datasets, by analogy with echo/antiecho processing in 2D correlation experiments, allows the dispersive components of the phase-twist lineshape to be cancelled and absorption mode peaks to be obtained. Unfortunately, the use of the ZS element entails a very significant sensitivity loss. This is particularly severe in second-order spin systems, which require the use of very narrow bandwidth, highly selective pulses in the ZS element.11 Such systems also give rise to unwanted responses in F1 that complicate analysis.
A number of methods for selective measurement of couplings in 2D J experiments have been reported, using either band-selective pulses or the ZS method, but these approaches are not broadband and/or suffer from significant sensitivity penalties.19 Very recently, Cotte and Jeannerat have reported a significant increase in the sensitivity of a ZS experiment, by using a selective pulse for simultaneous multi-slice irradiation, using non-equidistant phase modulation (nemoZS) to reduce the chances of accidental recoupling.20 However, this recovered sensitivity comes at the cost of increased spectral artefacts and experimental complexity.
Fortunately, the Pell–Keeler method can be used equally well with other J-refocusing sequence elements. We show here that an implementation combining the PSYCHE J-refocusing element with two extra swept-frequency chirp pulses (Fig. 1), to attenuate strong coupling responses,21 gives a major improvement in performance. The use of this triple spin echo (TSE-PSYCHE) 2D J sequence results in virtually artefact-free 2D J spectra with excellent sensitivity, absorption mode lineshapes, and no need for any special or non-linear post-processing. The new method can be used both for producing ultra-clean 1D pure shift spectra, by projecting the 2D spectrum at 45° in frequency space, and for extracting high resolution multiplet structure with minimal overlap. If all that is required is a 1D pure shift spectrum, applying the triple spin echo approach of Fig. 1 to the original interferogram 1D PSYCHE experiment, to give a 1D TSE-PSYCHE experiment (Fig. S2, ESI†), gives a significant improvement in spectral purity and artefact suppression (Fig. S3–S5, ESI†). The triple spin echo approach is very tolerant of B1 field inhomogeneity and pulse miscalibration, giving good results for RF power errors of up to 3 dB as evidenced by the 1D TSE-PSYCHE spectra of androst-4-ene-3,17-dione (Fig. S6, ESI†).
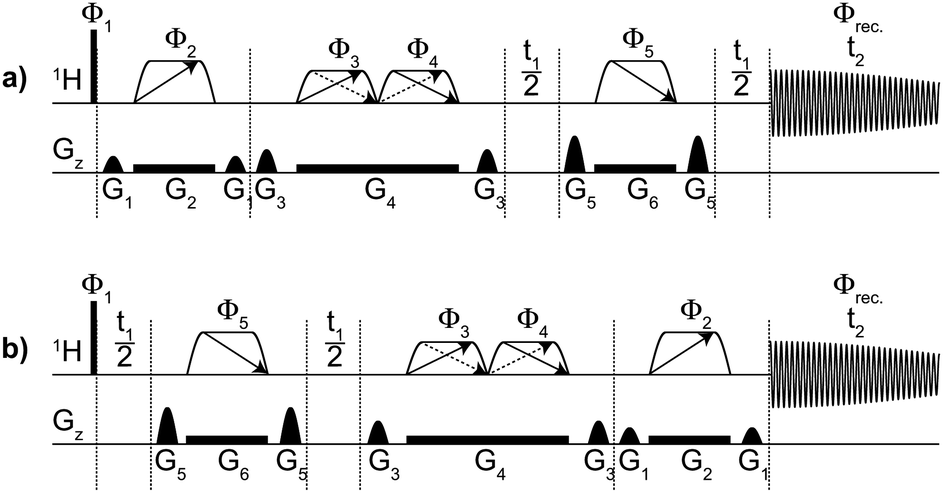 | ||
| Fig. 1 Pulse sequence for absorption mode TSE-PSYCHE 2D J spectroscopy. Independent datasets are acquired with (a) normal (N) and (b) J-reversed (R) sequences. The narrow rectangles denote a hard 90° RF pulse, the trapezoids with single arrows are 180° chirp pulses, and those with double arrows (saltires) are low flip angle chirp pulses (β = 15°) that sweep frequency simultaneously in opposite directions. G1 to G6 indicate field gradient pulses. The directions of frequency sweep for the first and last chirp pulses are opposed, in order to refocus the chemical shift evolution. Phase cycling: ϕ1 = x, −x, ϕ2 = x, ϕ3 = ϕ4 = x, x, y, y, ϕ5 = x, x, x, x, y, y, y, y and ϕrec. = x, −x, −x, x, −x, x, x, −x. | ||
The PSYCHE pulse sequence element achieves J refocusing by using a pair of low flip angle (β) pulses, analogous to those in the anti z-COSY22 experiment, that divide the available spins into two populations and manipulate these differentially. The PSYCHE element has already been used successfully in interferogram-based 1D and 2D pure shift experiments.11,12 For the PSYCHE pulse element, which consists of two chirp pulses, the active spins (those giving rise to the desired signal) are a proportion sin2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β and the passive spins (the remainder, including those coupled to the active spins) cos2β of the total. Thus, the sensitivity of PSYCHE experiment is determined by the sin2β term, and is normally about 5–10% of that of a conventional 1D proton measurement, while the corresponding figure for the ZS method is normally about 1%. The combination of frequency-swept (chirp) pulses and field gradients uses temporal and spatial averaging to suppress unwanted coherence transfer between spins through COSY22b and zero quantum23 pathways. This approach also significantly attenuates strong coupling21 artefacts. The active spins are refocused while the passive are left unperturbed, so the net effect is to refocus both chemical shifts and couplings.
β and the passive spins (the remainder, including those coupled to the active spins) cos2β of the total. Thus, the sensitivity of PSYCHE experiment is determined by the sin2β term, and is normally about 5–10% of that of a conventional 1D proton measurement, while the corresponding figure for the ZS method is normally about 1%. The combination of frequency-swept (chirp) pulses and field gradients uses temporal and spatial averaging to suppress unwanted coherence transfer between spins through COSY22b and zero quantum23 pathways. This approach also significantly attenuates strong coupling21 artefacts. The active spins are refocused while the passive are left unperturbed, so the net effect is to refocus both chemical shifts and couplings.
The one remaining source of unwanted signals is coherence transfer in which both coupled spins are flipped by the β pulses. For single-sweep pulses these signals have amplitudes proportional to sin4β, while the desired J-refocused signals are proportional to 2sin2βcos2β. The compromise between sensitivity and spectral purity is therefore simple to adjust in PSYCHE-based sequences, a low value of β increasing purity at the expense of sensitivity and a high value the reverse. Using chirp pulses that sweep frequency in opposite directions simultaneously (saltire pulses) changes these factors to 2sin4(β/2) and 4sin2(β/2)cos2(β/2) respectively, improving spectral purity (or sensitivity) by a further factor of four. The relationship between RF amplitude and flip angle of saltire chirp pulse is approximately linear, making calibration simple (Fig. S1, ESI†).
The PSYCHE-2D J pulse sequence of Fig. 1 is based on the classical hard pulse sequence 90° − t1/2 – 180° − t1/2 normally used for 2D J spectroscopy, but differs in two important respects. First, it follows Pell and Keeler in using a matched pair of pulse sequences in one of which (Fig. 1b) the PSYCHE sequence element comes after the evolution period t1, reversing the accumulated J evolution to give a reversed (R) dataset, and in the other (Fig. 1a) the PSYCHE element precedes t1 and gives a normal (N) dataset. PSYCHE has no effect on net J evolution in the sequence of Fig. 1a, being included solely to ensure that the same signal amplitudes result from both variants of the sequence. The N and R datasets can then be combined to cancel dispersive contributions and give pure 2D absorption mode lineshapes. The second difference from the usual sequence is that the hard pulse at the midpoint of t1 is replaced by a 180° chirp pulse under a field gradient, and a complementary chirp pulse is used at the end (a) or the beginning (b) of the sequence to refocus the chemical shift evolution (the order of the adjacent PSYCHE and 180° chirp elements in Fig. 1a and b is immaterial). Replacing the hard 180° pulse with the two spatially-resolved chirp pulses attenuates strong coupling artefacts just as in the sequence 1C described by Thrippleton, Edden and Keeler.21
Fig. 2 illustrates the application of the PSYCHE-2D J technique to 17β-estradiol, which has a crowded spectrum with a significant degree of strong coupling (Fig. 2a). In this spectrum, there are 7 signals in about a 200 Hz spectral width; most of them have 4 coupling partners, with J couplings ranging from 2 Hz to 13 Hz and an average multiplet width of 45 Hz (Fig. S9, ESI†). Fig. 2b shows the phase-sensitive 2D J spectrum, which has pure absorption mode lineshapes. The spectrum is of significantly improved quality with respect to sensitivity and level of artefacts compared to competing methods (Fig. S7, S11, and S12, ESI†). As the spectrum is almost devoid of artefacts, the application of symmetrisation, which can easily lead to misinterpretation of spectra, is unnecessary as it does not provide any significant further improvement. Projecting the 2D spectrum onto F2, as shown in Fig. 1b and 2b, gives access to a pure shift spectrum. (Since it is only the R type data obtained with sequence 1(b) that contribute to the pure shift signals in this projection,24 a pure shift spectrum with √2 better signal-to-noise ratio can be obtained by −45° projection of the simple 2D FT of these data.)
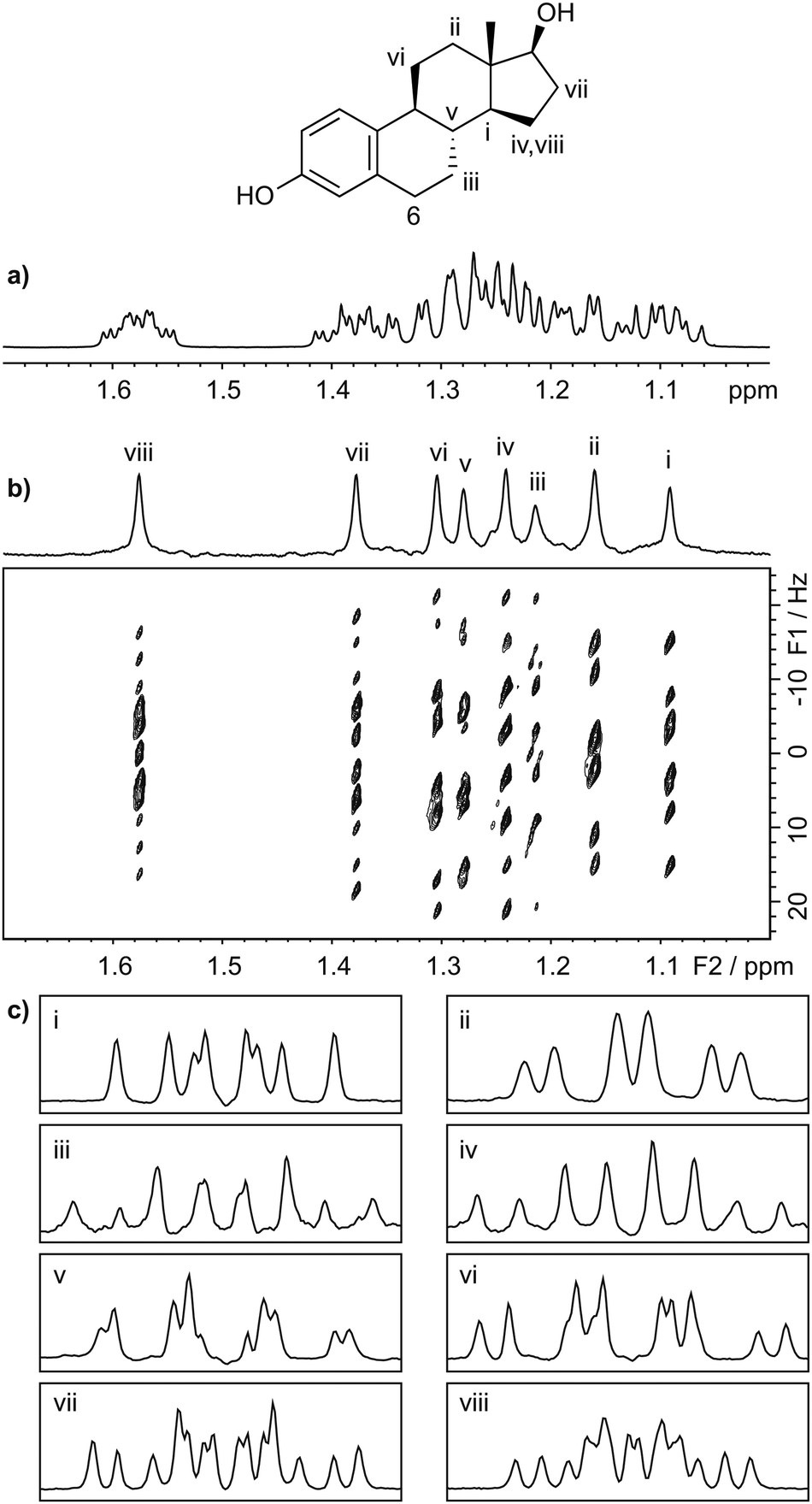 | ||
| Fig. 2 (a) 1D 1H spectrum of a sample of estradiol (scheme) in DMSO-d6, with (b) 45° tilted absorption mode 2D J spectrum acquired with the pulse sequence of Fig. 1, and (c) (i–viii) corresponding multiplet structures for signals, labelled on the pure shift F2 projection of (b), taken as vertical traces. | ||
The multiplet structure at each chemical shift is easily extracted from the complete 2D spectrum in the form of F1 traces, as shown in Fig. 2c. The chemical shift assignments and J values obtained from the 2D PSYCHE-2D J spectrum of 17β-estradiol (Fig. S9, ESI†) are largely in agreement with the literature,25 but reveal a few minor errors (confirmed by other measurements) in the published coupling constant values. Fig. 3 illustrates a second application of the PSYCHE-2D J technique, to the steroid hormone androst-4-ene-3,17-dione. This again shows that it is straightforward to extract high resolution multiplets from the absorption mode 2D J spectrum even where the 1D spectrum is too crowded for direct analysis. The improvement in spectral purity compared with the nemoZS method is illustrated in Fig. S3 and S12 of the ESI.† As comparison between Fig. 3 here and Fig. 4 of ref. 20 makes clear, PSYCHE-2D J also offers simpler data processing and analysis than nemoZS.
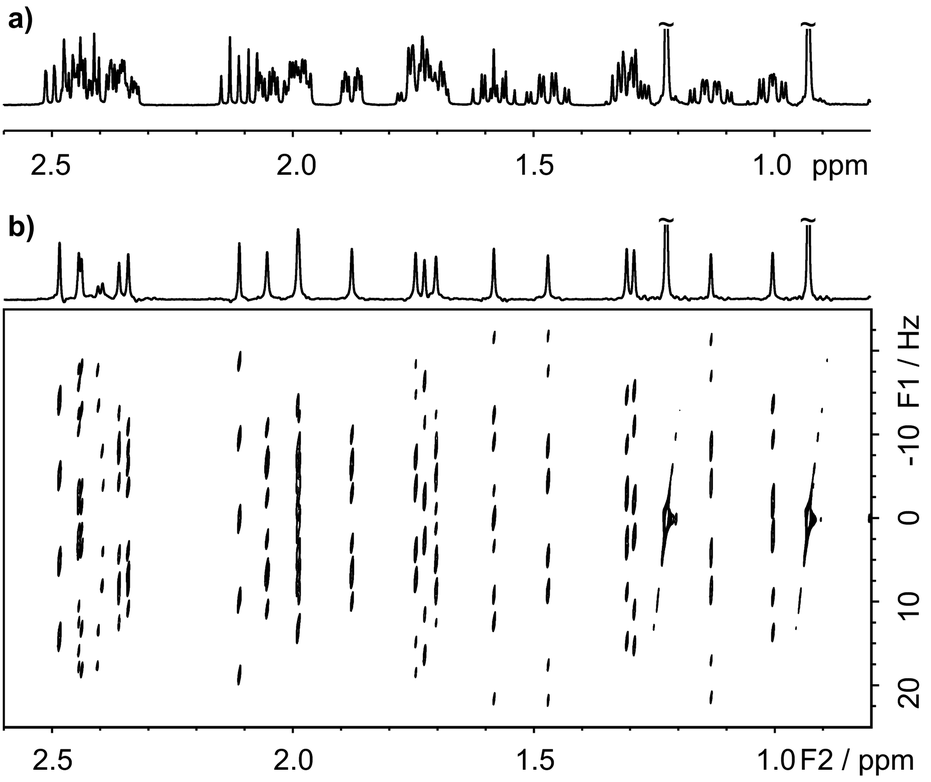 | ||
| Fig. 3 (a) 1D 1H spectrum of androst-4-ene-3,17-dione in chloroform-d, and (b) 45° tilted absorption mode 2D J spectrum acquired with the pulse sequence of Fig. 1, with (top) the pure shift spectrum obtained by projection of the tilted 2D spectrum onto F2. | ||
2D J spectroscopy was one of the first 2D NMR methods15 to be developed, but despite its potential in multiplet analysis has not found widespread use due to the limitations imposed by the phase-twist lineshape. Past attempts to circumvent these have suffered from artefacts and/or large sensitivity penalties. Here we present a clean, high sensitivity experiment that gives spectra with the desired double absorption mode lineshape, needs no special data processing, and is tolerant of pulse miscalibration. It opens up the possibility of using 2D J spectra for robust assignment and analysis of complex spectra, with resolution limited only by chemical shift difference and not by overlap between multiplets, a long-sought goal. Such spectra should greatly simplify both manual and automated spectral analysis, with applications throughout chemistry, biochemistry and structural biology.
This work was funded by the Engineering and Physical Sciences Research Council (grant numbers EP/L018500 and EP/M013820). Raw data for all the spectra shown, pulse sequence, parameters and shape files are freely available at DOI: 10.15127/1.266679. The authors thank Dr Peter Gierth of Bruker for assistance with RF calibration methods, and Dr Damien Jeannerat for MATLAB code for the generation of shaped pulses used in the nemoZS experiment.
References
- (a) R. W. Adams, eMagRes, John Wiley & Sons, Ltd., 2014 Search PubMed; (b) K. Zangger, Prog. Nucl. Magn. Reson. Spectrosc., 2015, 86–87, 1–20 CrossRef CAS.
- K. Zangger and H. Sterk, J. Magn. Reson., 1997, 124, 486–489 CrossRef CAS.
- J. R. Garbow, D. P. Weitekamp and A. Pines, Chem. Phys. Lett., 1982, 93, 504–509 CrossRef CAS.
- (a) J. A. Aguilar, S. Faulkner, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2010, 49, 3901–3903 CrossRef CAS; (b) J. A. Aguilar, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2011, 50, 9716–9717 CrossRef CAS; (c) N. Giraud, L. Béguin, J. Courtieu and D. Merlet, Angew. Chem., Int. Ed., 2010, 49, 3481–3484 CrossRef CAS; (d) A. Lupulescu, G. L. Olsen and L. Frydman, J. Magn. Reson., 2012, 218, 141–146 CrossRef CAS; (e) N. H. Meyer and K. Zangger, Angew. Chem., Int. Ed., 2013, 52, 7143–7146 CrossRef CAS.
- M. Nilsson and G. A. Morris, Chem. Commun., 2007, 933–935 RSC.
- G. A. Morris, J. A. Aguilar, R. Evans, S. Haiber and M. Nilsson, J. Am. Chem. Soc., 2010, 132, 12770–12772 CrossRef CAS.
- J. A. Aguilar, A. A. Colbourne, J. Cassani, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2012, 51, 6460–6463 CrossRef CAS.
- A. J. Pell and J. Keeler, J. Magn. Reson., 2007, 189, 293–299 CrossRef CAS.
- (a) L. Paudel, R. W. Adams, P. Király, J. A. Aguilar, M. Foroozandeh, M. J. Cliff, M. Nilsson, P. Sándor, J. P. Waltho and G. A. Morris, Angew. Chem., Int. Ed., 2013, 52, 11616–11619 CrossRef CAS; (b) P. Sakhaii, B. Haase and W. Bermel, J. Magn. Reson., 2009, 199, 192–198 CrossRef CAS.
- P. Kiraly, R. Adams, L. Paudel, M. Foroozandeh, J. Aguilar, I. Timári, M. Cliff, M. Nilsson, P. Sándor, G. Batta, J. Waltho, K. Kövér and G. Morris, J. Biomol. NMR, 2015, 62, 43–52 CrossRef CAS.
- M. Foroozandeh, R. W. Adams, N. J. Meharry, D. Jeannerat, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2014, 53, 6990–6992 CrossRef CAS.
- M. Foroozandeh, R. W. Adams, M. Nilsson and G. A. Morris, J. Am. Chem. Soc., 2014, 136, 11867–11869 CrossRef CAS.
- (a) R. W. Adams, L. Byrne, P. Kiraly, M. Foroozandeh, L. Paudel, M. Nilsson, J. Clayden and G. A. Morris, Chem. Commun., 2014, 50, 2512–2514 RSC; (b) L. Castañar, P. Nolis, A. Virgili and T. Parella, Chem. – Eur. J., 2013, 19, 17283–17286 CrossRef; (c) J. Ying, J. Roche and A. Bax, J. Magn. Reson., 2014, 241, 97–102 CrossRef CAS.
- S. Islam, J. A. Aguilar, M. W. Powner, M. Nilsson, G. A. Morris and J. D. Sutherland, Chem. – Eur. J., 2013, 19, 4586–4595 CrossRef CAS.
- W. P. Aue, J. Karhan and R. R. Ernst, J. Chem. Phys., 1976, 64, 4226–4227 CrossRef CAS.
- S. Simova, H. Sengstschmid and R. Freeman, J. Magn. Reson., 1997, 124, 104–121 CrossRef CAS.
- A. Bax, A. F. Mehlkopf and J. Smidt, J. Magn. Reson., 1980, 40, 213–219 CAS.
- (a) V. A. Mandelshtam, Q. N. Van and A. J. Shaka, J. Am. Chem. Soc., 1998, 120, 12161–12162 CrossRef CAS; (b) P. Mutzenhardt, F. Guenneau and D. Canet, J. Magn. Reson., 1999, 141, 312–321 CrossRef CAS; (c) J.-M. Nuzillard, J. Magn. Reson., Ser. A, 1996, 118, 132–135 CrossRef CAS.
- (a) A. Verma and B. Baishya, ChemPhysChem, 2015, 16, 2687–2691 CrossRef CAS; (b) D. Pitoux, B. Plainchont, D. Merlet, Z. Hu, D. Bonnaffé, J. Farjon and N. Giraud, Chem. – Eur. J., 2015, 21, 9044–9047 CrossRef CAS; (c) J. E. Herbert Pucheta, D. Pitoux, C. M. Grison, S. Robin, D. Merlet, D. J. Aitken, N. Giraud and J. Farjon, Chem. Commun., 2015, 51, 7939–7942 RSC.
- A. Cotte and D. Jeannerat, Angew. Chem., Int. Ed., 2015, 54, 6016–6018 CrossRef CAS.
- M. J. Thrippleton, R. A. E. Edden and J. Keeler, J. Magn. Reson., 2005, 174, 97–109 CrossRef CAS.
- (a) H. Oschkinat, A. Pastore, P. Pfändler and G. Bodenhausen, J. Magn. Reson., 1986, 69, 559–566 CAS; (b) A. J. Pell, R. A. E. Edden and J. Keeler, Magn. Reson. Chem., 2007, 45, 296–316 CrossRef CAS.
- M. J. Thrippleton and J. Keeler, Angew. Chem., Int. Ed., 2003, 42, 3938–3941 CrossRef CAS.
- K. Nagayama, P. Bachmann, K. Wüthrich and R. R. Ernst, J. Magn. Reson., 1978, 31, 133–148 CAS.
- J. Guo, R. I. Duclos Jr, V. K. Vemuri and A. Makriyannis, Tetrahedron Lett., 2010, 51, 3465–3469 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; description of saltire chirp pulses and related calculations; multiplet structures and corresponding J values obtained using PSYCHE-2D J for 17β-estradiol; experimental data comparing the various methods discussed for different samples; and Bruker pulse sequence codes for modified interferogram PSYCHE, new TSE-PSYCHE with internal RF field calculations, and PSYCHE-2D J. See DOI: 10.1039/c5cc06293d |
| This journal is © The Royal Society of Chemistry 2015 |