
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrophilic non-precious metal nitrogen-doped carbon electrocatalysts for enhanced efficiency in oxygen reduction reaction†
Guang-Ping
Hao
a,
Nastaran Ranjbar
Sahraie
b,
Qiang
Zhang
c,
Simon
Krause
a,
Martin
Oschatz
a,
Alicja
Bachmatiuk
de,
Peter
Strasser
*b and
Stefan
Kaskel
*af
aDepartment of Inorganic Chemistry, Technische Universität Dresden, Bergstraße 66, 01069 Dresden, Germany. E-mail: Stefan.Kaskel@chemie.tu-dresden.de
bThe Electrochemical Energy, Catalysis, and Materials Science Laboratory, Department of Chemistry, Chemical Engineering Division, Technical University Berlin, Straße des 17. Juni 124, 10623 Berlin, Germany. E-mail: pstrasser@tu-berlin.de
cBeijing Key Laboratory of Green Chemical Reaction Engineering and Technology, Department of Chemical Engineering, Tsinghua University, Beijing 100084, P. R. China
dIFW Dresden, Institute of Complex Materials, P. O. Box 270116, D-01171, Dresden, Germany
eCenter of Polymer and Carbon Materials, Polish Academy of Sciences, M. Curie-Sklodowskiej 34, Zabrze 41-819, Poland
fFraunhofer Institute for Material and Beam Technology, Winterbergstraße 28, 01277, Dresden, Germany
First published on 5th October 2015
Abstract
Exploring the role of surface hydrophilicity of non-precious metal N-doped carbon electrocatalysts in electrocatalysis is challenging. Herein we discover an ultra-hydrophilic non-precious carbon electrocatalyst, showing enhanced catalysis efficiency on both gravimetric and areal basis for oxygen reduction reaction due to a high dispersion of active centres.
The oxygen reduction reaction (ORR) is a fundamental electrochemical reaction for fuel cells and metal-air batteries. ORR research has long been focused on the development and understanding of new non-precious nitrogen-doped carbon catalysts that result in significant cost reduction by platinum metal substitution. To catalyze broader commercialization of such devices and technologies, efficient and affordable non-precious electrocatalysts will ultimately be required. One of the most prominent examples is pyrolyzed solid materials consisting of non-precious metal/nitrogen/carbon (M/N/C-) composites,1 or metal-free, heteroatom-doped nanocarbons.2 The nature of the active sites in terms of the modulation of electron donating/withdrawing capability of the carbon basal plane by incorporated heteroatoms of the M/N/C electrocatalysts has been under intensive investigation and has become more and more clear.3 However, a detailed understanding of the effect of surface hydrophilicity and wettability on the dispersion of metal-related active sites as well as their effects on the catalysis efficiency of the M/N/C materials has remained elusive.
On the one hand, a hydrophilic pore surface benefits a facile loading and a high dispersion of active metal-related species, because hydrophilic affinity can be created between hydrophilic pore walls and precursors, and further inhibits precursor random migration and agglomeration.4 On the other hand, a hydrophilic pore surface may also influence the transport of hydrated O2 to the electrochemically active centres under hydrated conditions, finally affecting the activity.5 To our knowledge, such effects stemming from surface hydrophilicity have been rarely investigated to date.
Taking these into consideration, in this contribution, we surface engineered a number of different carbon-based materials with surface characteristics ranging from an ultra-hydrophilic carbon network to an ultra-hydrophobic carbon black. We observe that hydrophilicity, quantified by water adsorption isotherms at 298 K, is correlated with much enhanced ORR catalysis efficiency.
Firstly, a group of ultra-hydrophilic electrocatalysts (Fe/N_1/3.2, Fe/Cu/N_1.3/1/8, and Cu/N_1/4, according to the nominal atomic ratio of Fe/N, Fe/Cu/N and Cu/N in the synthesis, Fig. 1a–c) were fabricated by a facile and scalable impregnation and subsequent pyrolysis and leaching method (Experimental section, ESI†) based on an unprecedentedly hydrophilic carbon network (DUT-110, DUT = Dresden University of Technology, derived from a functional complex6). The highly hydrophilic surface property of DUT-110 was confirmed by the sharp uptake in the water vapor adsorption isotherm from the very beginning, showing a record value until P/P0 < 0.3 (Fig. S1a, ESI†).7 The narrow and rich micropores were proven by the type I isotherm and pore size distribution based on N2 physisorption data (Fig. S1b, ESI†) as well as high resolution TEM images (Fig. S1c, ESI†); while the STEM mapping and XPS spectrum revealed the highly heteroatom-doped feature, with a surface composition of C, N, and O with the atomic content of 74.7, 14.3, and 10.33%, respectively (Fig. S1d and e, ESI†). In parallel, hydrophobic non-precious electrocatalysts were prepared under the same principle through the modification of N-containing polymers (polyaniline, PANI or N-containing ionic liquid, N,N-ethyl-methyl-imidazolium–dicyanamide) and FeCl3, but based on highly hydrophobic carbon black (Ketjen EC 600J). Note that the pyrolysis and leaching treatment as well as the following performance evaluation were kept identical with those of the hydrophilic groups.
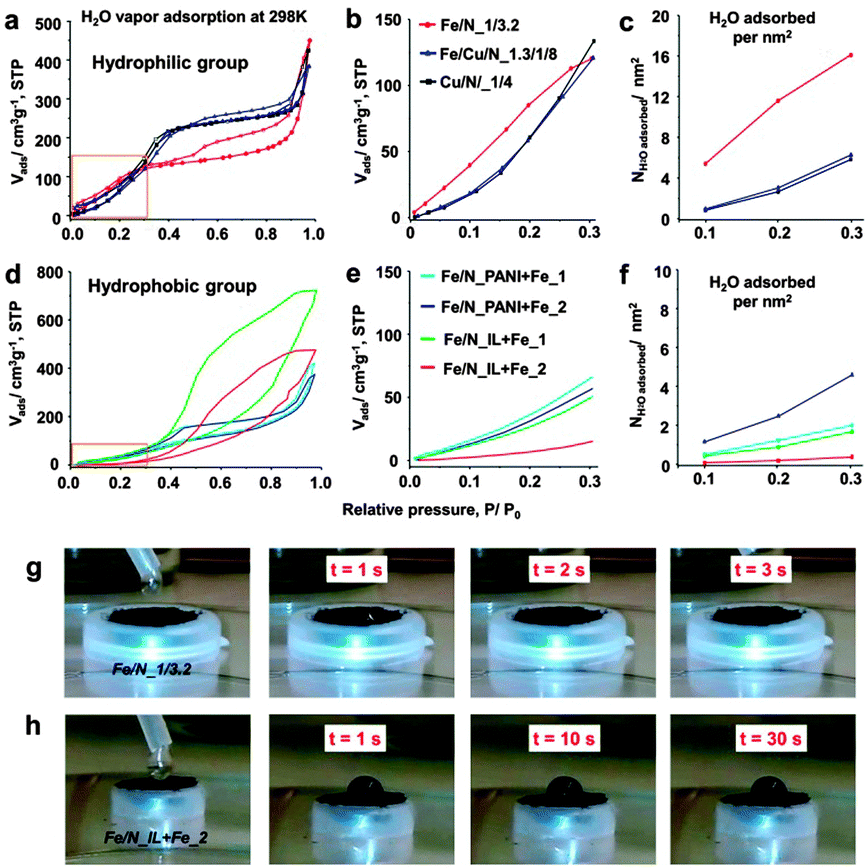 | ||
| Fig. 1 H2O vapor adsorption isotherm at different relative pressure range at 298 K of hydrophilic groups (a–c) and hydrophobic groups (d–f). (g and h) Comparison of dynamic water contact angle changes of typical samples of the two groups. | ||
Surprisingly, the final hydrophilic electrocatalysts, Fe/N_1/3.2, Fe/Cu/N_1.3/1/8, and Cu/N_1/4, maintained largely the high surface hydrophilicity. They all exhibit relatively good hydrophilic properties, with the water uptake of 120.4, 120.5, and 133.6 cm3 g−1 (equivalent to 5.37, 5.38, and 5.96 mmol g−1) at P/P0 = 0.3 for Fe/N_1/3.2, Fe/Cu/N_1.3/1/8, and Cu/N_1/4, respectively (Fig. 1a). The water adsorption behavior in the range of 0 < P/P0 < 0.3 is mainly determined by the surface hydrophilicity;7 thus, we further compared the water sorption uptake in this pressure range (Fig. 1b). All the hydrophilic samples, i.e., Fe/N_1/3.2, Fe/Cu/N_1.3/1/8, and Cu/N_1/4, exhibit a higher water adsorption uptake, even though their less developed porosity and low surface area (200–574 m2 g−1, Fig. S2, ESI†) were comparable to hydrophobic materials (340–1079 m2 g−1, Table S1, ESI†). After normalization to specific surface area (Fig. 1c), the hydrophilicity order is Fe/N_1/3.2 > Fe/Cu/N_1.3/1/8 > Cu/N_1/4. For instance, the water adsorption uptake is calculated to be 16.2 H2O molecules per nm2, 6.7 H2O molecules per nm2 and 6.2 H2O molecules per nm2 at P/P0 = 0.3 for Fe/N_1/3.2, Fe/Cu/N_1.3/1/8, and Cu/N_1/4, respectively. However, the water adsorption isotherm of the hydrophobic electrocatalysts displays much lower water adsorption uptakes, indicating a much lower surface hydrophilicity (Fig. 1d–f). To illustrate the difference in hydrophilicity vividly, the water contact angles were recorded dynamically after water droplets contacted the carbon pellet and were compared (herein we show the samples of Fe/N_1/3.2 and Fe/N_IL + Fe_2 in Fig. 1g and h). For hydrophilic Fe/N_1/3.2, the water droplet can be adsorbed in 3 s with a final contact angle of ca. 0, while the sample of Fe/N_IL + Fe_2 was not wetted until 30 s, again confirming the distinct surface hydrophilicity. Morphologically, all the hydrophilic electrocatalysts exhibit a highly interconnected network structure (Fig. S3, ESI†), but a much denser structure compared with their host carbon networks DUT-110 (Fig. S1d, ESI†). This is due to the shrinkage of the carbon skeletons during high temperature pyrolysis.
Based on the unique hydrophilicity and narrow micropores, a high dispersion of metal-related nanoparticles was expected for hydrophilic electrocatalysts. Thus, the elemental distribution was analysed by STEM images and elemental maps as well as dark-field TEM images (Fig. 2). For Fe/N_1/3.2 (Fig. 2a), large and bright particles can be observed, while for Fe/Cu/N_1.3/1/8, only very few isolated particles can be detected (Fig. 2b). This observation is consistent with the TEM images (Fig. S4, ESI†). Importantly, except some large particles, the distribution of small metal species is very homogeneous, indicating highly dispersed nanoclusters or even mononuclear metal species embedded in the carbon matrix. The dark-field TEM images (Fig. 2e and f), corresponding to their relevant TEM images (Fig. 2c and d), again confirmed a high and uniform distribution of metal-related nanoclusters over the whole framework. The metal content determined by the ICP technique is 3.8 wt% Fe for Fe/N_1/3.2 and 1.22 wt% Fe for Fe/N_1/3.2. Note that these metal nanoclusters should be tightly embedded in the carbon matrix, since all these electrocatalysts have been extensively leached in 2.0 M H2SO4 at 110 °C for 24 h before harvesting for characterisation and application. Furthermore, the hybrid structure composed of graphitic domains and amorphous carbons was revealed by Raman spectra (Fig. S5, fitting details in Table S2, ESI†) with the ID/IG ratio of 1.55, 1.82 and 2.11 for Fe/N_1/3.2, Fe/Cu/N_1.3/1/8 and Cu/N_1/4, respectively, confirming the observation by TEM images (Fig. S4, ESI†).
 | ||
| Fig. 2 STEM maps of Fe/N_1/3.2 (a) and Fe/Cu/N_1.3/1/8 (b), TEM image and the corresponding dark-field TEM images of Fe/N_1/3.2 (c and e) and Fe/Cu/N_1.3/1/8 (d and f). | ||
For non-metal elements such as C, O, and N, a homogeneous dispersion was also observed by elemental maps (Fig. 2a and b), indicating a uniform doped structure. Interestingly, comparing the Fe or Cu maps with O maps, a strong correlation is found between the metal and O, particularly for large particles, indicating their oxide phase in nature. This observation is further confirmed by their XPS analysis (Fig. S6, ESI†). Furthermore, Table S1 (ESI†) also lists other structural parameters such as the specific surface area analyzed by N2 adsorption, the surface non-metal compositions determined by XPS and the metal species detected by ICP for all the hydrophilic and hydrophobic catalyst groups in order to obtain a reliable correlation between the structural parameters and the subsequent catalysis performance.
We first evaluated the ORR activity of the hydrophilic group, i.e., Fe/N_1/3.2, Fe/Cu/N_1.3/1/8, and Cu/N_1/4. Linear sweep voltammetry (LSV, Fig. 3a) in 0.10 M KOH was employed to investigate the catalytic activity of the catalysts compared to Pt/C benchmark catalysts. The onset potentials (Eonset, a noteworthy onset potential is defined as the potential at which the current density reaches 1.0 mA cm−2) are 0.90, 0.92, and 0.89 V for Fe/N_1/3.2, Fe/Cu/N_1.3/1/8, and Cu/N_1/4, respectively (Fig. 3a, Table S1, ESI†). The Eonset of Fe/Cu/N_1.3/1/8 is positive and comparable with those of the reported state-of-the-art non-precious catalysts such as Fe/N-doped nanocarbons (e.g. N-CNT/Fe3C, N-doped carbon nanoplate/Fe3C, Fe@Fe3C/N-doped carbon),8 Fe and/or N-doped porous carbons with higher surface area or larger pores such as mesopores9 or hierarchical pores,10 and the hybrid N-Fe-CNT/carbon nanoparticle with higher Fe content.11 The half-wave potential (E1/2) shows a similar trend that is also comparable with that of the state-of-the-art non-precious electrocatalysts (Table S3, ESI†).9–12 Particularly, the high activity reflected by the positive Eonset and E1/2 of Fe/Cu/N_1.3/1/8 originates from the highly dispersed active sites and highly accessible porosity.
 | ||
| Fig. 3 ORR catalysis evaluation in O2-saturated 0.10 M KOH. (a) Linear sweep voltammetry (LSV) ORR plots under conditions of room temperature, at a rotation speed of 1500 rpm, scan rate of 10 mV s−1, the non-precious catalysts loading of 0.80 mg cm−2, and the benchmark Pt loading of 10 μg cm−2. (b) The relationship between hydrophilicity in terms of water molecules adsorbed per nm2 based on water adsorption data at P/P0 = 0.3 and mass activity. (c) Structure–performance comparison including surface hydrophilicity, nitrogen content (atomic%, by XPS) and Fe content (wt%, by ICP) with mass activity in ORR electrocatalysis under identical conditions. | ||
The mass activity indicates the utilization efficiency of catalysts on a gravimetric basis. For the hydrophilic series, relatively higher mass activities up to 413.3, 232.5, and 137.7 mAmg−1 were calculated for Fe/N_1/3.2, Fe/Cu/N_1.3/1/8, and Cu/N_1/4, respectively (Fig. 3b). For the hydrophobic samples, the mass activity is one order of magnitude lower (Fig. 3b). The much higher mass activity of the hydrophilic samples originated from the positively shifted onset and half-wave potentials, indicating the large density of accessible active sites due to the high dispersion of electrochemically active sites benefiting from the highly hydrophilic carbon surface. In order to understand quantitatively, we further normalized the mass activity by the surface area. The obtained specific activities are 2067, 482.4, and 239.9 mA m−2 for Fe/N_1/3.2, Fe/Cu/N_1.3/1/8, and Cu/N_1/4, respectively. This trend is also consistent with that of mass activity, reflecting the remarkably high surface efficiency (Fig. S7 and Table S1, ESI†).
Moreover, we correlated the Fe and N content, and the surface area as well as surface hydrophilicity with mass activity. However, it is difficult to find a clear trend between either mass or specific activity and doping properties (Fig. 3c, black and grey lines, Fig. S8, ESI†) or specific surface area (Table S1 and Fig. S9, ESI†). In contrast, a clear correlation between mass activity and surface hydrophilicity was observed. This is probably because (1) the higher surface hydrophilicity induces a higher dispersion of active sites, and (2) hydrophilic pores benefit an easy accessibility to the active sites of reactants (such as hydrated O2 as ORR proceeds). Besides the comparison between specific samples, a comparison between the hydrophobic group and the ultra-hydrophilic group confirmed the same rule (Fig. 3c, red trend line).
The high dispersion of active sites benefiting from surface hydrophilicity has been proven above by STEM mapping and dark field TEM images. However, to observe the effect of hydrophilicity on the diffusion of hydrated O2 and resultant H2O is challenging. For the diffusion of hydrated O2 near reaction interfaces, high surface hydrophilicity may be beneficial.5b–d In order to explain this point, we hypothesized a “physical structure” (Fig. S10, ESI†), where water molecules around hydrated O2 molecules can be readily stripped by the hydrophilic micropore walls when approaching the carbon slit pores, then the liberated O2 molecules can freely diffuse to the active sites and thus can accelerate the reaction. The O2 adsorption was investigated for the active hydrophilic group, which can give the first clue that high surface hydrophilicity enhanced the O2 adsorption (Fig. S11, ESI†). All hydrophilic samples exhibit a combined type I isotherm (Fig. S9a and b ESI†), indicating a strong interaction between O2 molecules and doped carbon pore walls. After normalization by the specific surface area, the samples showed a higher areal uptake, indicating the preferential O2 adsorption and high surface utilization efficiency for trapping O2 molecules (Fig. S11c, ESI†). Interestingly, the O2 capture behaviour is consistent with that of water sorption in the same pressure range.
In addition, one would realize that the high surface hydrophilicity of the catalysts may also cause a delay of water desorption when used in applications that generate water such as alkaline fuel cells (AFCs). In this case, further H2 reduction can be applied to effectively reduce the surface hydrophilicity (Fig. S12, ESI†). Through this way, the possible flooding issues can be avoided in potential applications such as AFCs.
In summary, exemplified using the non-precious carbon based ORR electrocatalyst concept, we surface engineered a number of different carbon based materials with surface characteristics ranging from an ultra-hydrophilic carbon network to ultra-hydrophobic carbon black. A high surface hydrophilicity has been found to give an easily wetted surface which first ensures a high dispersion of metal-related active sites and may also increase the accessibility of reactants to active centres, and thus may increase the surface and mass utilization efficiency of catalysts. This work provides fresh insight into the controlling material parameters of non-precious ORR catalysts, and as such offers new clues and strategies on how to increase the ORR catalysis efficiency by tuning the surface chemistry of non-precious electrocatalysts. The insight into hydrophilicity may also be important to other heterogeneous catalytic reactions such as CO2 electro-reduction, glucose oxidation, and metal–air batteries, catalyzed on hydrated carbon surfaces.
We thank Prof A. Eychmüller and S. Klosz for Raman spectra measurement, Dr I. Senkovska for help in the O2 adsorption measurement. G.-P. H. acknowledges the financial support from the Alexander von Humboldt Foundation.
Notes and references
- (a) M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71 CrossRef PubMed; (b) F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114 RSC; (c) G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443 CrossRef CAS PubMed; (d) M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed; (e) K. Wood, R. O'Hayre and S. Pylypenko, Energy Environ. Sci., 2014, 7, 1212 RSC; (f) D.-W. Wang and D. Su, Energy Environ. Sci., 2014, 7, 576 RSC; (g) G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878 CrossRef CAS PubMed.
- (a) K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed; (b) L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321 CrossRef CAS PubMed; (c) W. Wei, H. Liang, K. Parvez, X. Zhuang, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2014, 53, 1570 CrossRef CAS PubMed; (d) Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 4394 CrossRef CAS PubMed; (e) J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496 CrossRef CAS PubMed; (f) D. Yu, Q. Zhang and L. Dai, J. Am. Chem. Soc., 2010, 132, 15127 CrossRef CAS PubMed.
- (a) J.-L. Shui, N. K. Karan, M. Balasubramanian, S.-Y. Li and D.-J. Liu, J. Am. Chem. Soc., 2012, 134, 16654 CrossRef CAS PubMed; (b) N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, J. Am. Chem. Soc., 2013, 135, 15443 CrossRef CAS PubMed; (c) Y. Zhu, B. Zhang, X. Liu, D.-W. Wang and D. Su, Angew. Chem., Int. Ed., 2014, 53, 10673 CrossRef CAS PubMed; (d) U. I. Kramm, M. Lefèvre, N. Larouche, D. Schmeisser and J.-P. Dodelet, J. Am. Chem. Soc., 2014, 136, 978 CrossRef CAS PubMed; (e) C. H. Choi, H.-K. Lim, M. W. Chung, J. C. Park, H. Shin, H. Kim and S. I. Woo, J. Am. Chem. Soc., 2014, 136, 9070 CrossRef CAS PubMed.
- (a) A.-H. Lu and F. Schüth, Adv. Mater., 2006, 18, 1793 CrossRef CAS; (b) H. Yang and D. Zhao, J. Mater. Chem., 2005, 15, 1217 CAS; (c) Q. Wang, Z.-Y. Zhou, Y.-J. Lai, Y. You, J.-G. Liu, X.-L. Wu, E. Terefe, C. Chen, L. Song, M. Rauf, N. Tian and S.-G. Sun, J. Am. Chem. Soc., 2014, 136, 10882 CrossRef CAS PubMed.
- (a) S. Holdcroft, Chem. Mater., 2014, 26, 381 CrossRef CAS; (b) D. Yu, E. Nagelli, F. Du and L. Dai, J. Phys. Chem. Lett., 2010, 1, 2165 CrossRef CAS; (c) G. J. Sohn, H. J. Choi, I. Y. Jeon, D. W. Chang, L. Dai and J. B. Baek, ACS Nano, 2012, 6, 6345 CrossRef CAS PubMed; (d) I. Y. Jeon, H. J. Choi, S. M. Jung, J. M. Seo, M. J. Kim, L. Dai and J. B. Baek, J. Am. Chem. Soc., 2013, 135, 1386 CrossRef CAS PubMed.
- (a) G.-P. Hao, G. Mondin, Z. Zheng, T. Biemelt, S. Klosz, R. Schubel, A. Eychmüller and S. Kaskel, Angew. Chem., Int. Ed., 2015, 54, 1941 CrossRef CAS PubMed; (b) K. Kaneko, Nat. Chem., 2015, 7, 194 CrossRef CAS PubMed.
- (a) D. D. Do, S. Junpirom and H. D. Do, Carbon, 2009, 47, 1466 CrossRef CAS; (b) T. Matsuoka, H. Hatori, M. Kodama, J. Yamashita and N. Miyajima, Carbon, 2004, 42, 2329 CrossRef; (c) J. K. Brennan, K. T. Thomson and K. E. Gubbins, Langmuir, 2002, 18, 5438 CrossRef CAS; (d) H.-J. Wang, A. Kleinhammes, T. P. McNicholas, J. Liu and Y. J. Wu, J. Phys. Chem. C, 2014, 118, 8474 CrossRef CAS; (e) Y. Tao, M. Endo and K. Kaneko, J. Am. Chem. Soc., 2009, 131, 904 CrossRef CAS PubMed; (f) T. Ohba and K. Kaneko, J. Phys.: Conf. Ser., 2009, 177, 012001 CrossRef.
- (a) W. Yang, X. Liu, X. Yue, J. Jia and S. Guo, J. Am. Chem. Soc., 2015, 137, 1436 CrossRef CAS PubMed; (b) S. Zhao, H. Yin, L. Du, L. He, K. Zhao, L. Chang, G. Yin, H. Zhao, S. Liu and Z. Tang, ACS Nano, 2014, 8, 12660 CrossRef CAS PubMed; (c) K. Ai, Y. Liu, C. Ruan, H. Lu and G. Lu, Adv. Mater., 2013, 25, 998 CrossRef CAS PubMed.
- (a) Z. Li, G. Li, L. Jiang, J. Li, G. Sun, C. Xia and F. Li, Angew. Chem., Int. Ed., 2015, 54, 1494 CrossRef CAS PubMed; (b) R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565 CrossRef CAS PubMed.
- (a) H.-W. Liang, X. Zhuang, S. Brüller, X. Feng and K. Müllen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed; (b) G.-L. Tian, M.-Q. Zhao, D. Yu, X.-Y. Kong, J.-Q. Huang, Q. Zhang and F. Wei, Small, 2014, 10, 2251 CrossRef CAS PubMed.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed.
- (a) S. Zhao, H. Yin, L. Du, L. He, K. Zhao, L. Chang, G. Yin, H. Zhao, S. Liu and Z. Tang, ACS Nano, 2014, 8, 12660 CrossRef CAS PubMed; (b) W. Yang, X. Liu, X. Yu, J. Jia and S. Guo, J. Am. Chem. Soc., 2015, 137, 1436 CrossRef CAS PubMed; (c) N. R. Sahraie, J. P. Paraknowitsch, C. Göbel, A. Thomas and P. Strasser, J. Am. Chem. Soc., 2014, 136, 14486 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc06256j |
| This journal is © The Royal Society of Chemistry 2015 |