
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA compact planar low-energy-gap molecule with a donor–acceptor–donor nature based on a bimetal dithiolene complex†
Mikihiro
Hayashi
*a,
Kazuya
Otsubo
a,
Tatsuhisa
Kato
b,
Kunihisa
Sugimoto
c,
Akihiko
Fujiwara
d and
Hiroshi
Kitagawa
*ae
aDivision of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa Oiwake-cho, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: mhayashi@kuchem.kyoto-u.ac.jp; kitagawa@kuchem.kyoto-u.ac.jp
bInstitute for the Promotion of Excellence in Higher Education, Kyoto University, Yoshida-Nihonmatsu, Sakyo-ku, Kyoto 606-8501, Japan
cJapan Synchrotron Radiation Research Institute (JASRI), SPring-8, 1-1-1 Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5198, Japan
dDepartment of Nanotechnology for Sustainable Energy, Graduate School of Science and Technology, Kansei Gakuin University, Gakuen, Sanda, Hyogo 669-1337, Japan
eCore Research for Evolutional Science and Technology (CREST), Japan Science and Technology Agency (JST), 7 Goban-cho, Chiyoda-ku, Tokyo 102-0075, Japan
First published on 4th September 2015
Abstract
We present the first report of a compact, planar and low-energy-gap molecule based on a π-conjugated bimetal system comprising a tetrathiooxalate (tto) skeleton. The observed low HOMO–LUMO energy gap (1.19 eV) is attributed to its donor–acceptor–donor (D–A–D) nature because the tto skeleton acts as an electron acceptor as well as a tiny and noninnocent bridging moiety.
For the last half century, electron donor (D) and acceptor (A) molecules have been intensively developed because their intermolecular charge-transfer interactions play important roles in optical and transport properties, such as electron transfer in photosynthesis or carrier generation in charge-transfer complexes.1 Lately, the direct connection of organic D–A molecules through π-conjugation has been investigated because of the molecules’ intramolecular interactions. This feature affords characteristic properties such as strong photoabsorption and intense emission in the visible region because of the small energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).2 In addition to the D–A type, D–A–D or A–D–A-type molecules have also been developed, in which versatile electrochemical properties such as multiredox behavior and mixed valence states, as well as the intramolecular D–A interactions are observed. Such D–A–D or A–D–A systems have been applied to processes involving light energy, such as photoelectric conversion or molecular optical switching devices.3 In terms of materials for solar conversion, molecules that can harvest light in the near infrared (NIR) region are desirable because NIR radiation accounts for a large portion of the spectrum of sunlight. Large π-extended structures such as porphyrins and fullerenes have been explored for their NIR absorption characteristics,4 but while such systems are promising, small, flat D–A–D or A–D–A molecules are expected to be advantageous for the facile fabrication of molecular devices.5 However, there have been no reports of such compact and planar D–A–D or A–D–A molecules with low-energy-gap characteristics.
Recently, organic–inorganic hybrid materials with multimetal centers have been studied extensively because of their unique multiredox behavior.6 The square planar metal dithiolene complex is a typical example because dithilene ligands have a noninnocent nature based on delocalization of the electron density across the whole metal dithiolene ring in addition to a strong interaction between the d-orbitals of the metal atom and the p-orbitals of the sulfur atoms.7 Among the many kinds of dithiolene ligands, tetrathiooxalate (tto) has potential to be the smallest bridging component of π-conjugated bimetal complexes which show a multiredox behavior. Therefore, tto-bridged bimetal complexes are candidates for compact and planar D–A–D or A–D–A molecules; however, there are few reports because of the difficulty of synthesis.8 In this paper, we attempt to expose the D–A–D nature of a compact, planar, low-energy-gap tto-bridged bimetal system and show the previously unrevealed ability of the tto skeleton to act as an electron acceptor.
To achieve this aim, we designed a new bimetal complex: a tto-bridged dinuclear Ni dithiolene complex with a pyradinedithiole (pdt) ligand ([(tto)Ni(pdt)2]2−). We adopted a synthetic method to avoid the oligomerization reaction of [Ni(tto)]n in the bimetallic form by adding terminal ligands (Scheme 1).9 Careful addition of the pdt ligand afforded [(tto)Ni(pdt)2]2− with two tetrabutylammonium ions (TBA) as countercations (TBA2[(tto)Ni(pdt)2]: complex 1), with a small synthetic yield (3.4%). According to this synthetic route, TBA2[Ni(pdt)2] was also obtained with a high yield (30%) due to the high coordination abilities of both tto and pdt ligands. Single-crystal X-ray structural analysis, 1H NMR and elemental analysis were utilized for characterization (see ESI†).
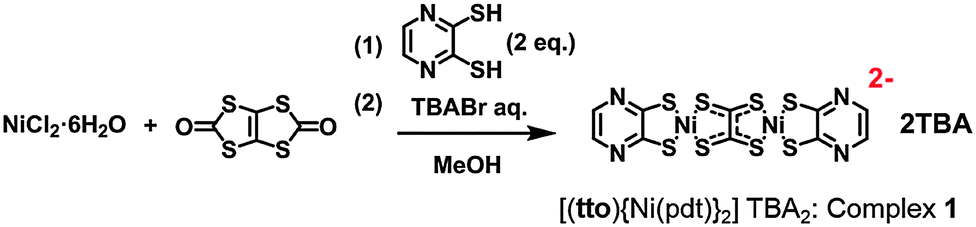 | ||
| Scheme 1 Synthesis of [(tto)Ni(pdt)2]TBA2 (complex 1). | ||
Single crystals were grown by a diffusion method (dimethylsulfoxide (DMSO)/diethylether) and the crystal structure of complex 1 determined at 300 K is shown in Fig. 1a. Some of the DMSO solvent was included in the crystal giving a formula of 1·2DMSO. In view of the molecular geometry, complex 1 has a flat structure that is attributed to full π-conjugation at 300 K. It is known that the tto moiety can act as either a dianionic or tetraanionic counteranion, which has been structurally identified by its central CC bond length.10 The CC bond length of complex 1 (C(1)–C(2)) was 1.415(8) Å at 300 K which indicates a single bond rather than a double bond, so that the tto moiety has a dianionic rather than tetraanionic character. Resonance Raman spectroscopy reveals that complex 1 exhibits an intense peak at around 1130 cm−1 assigned to the stretching vibration of the tto skeleton. This peak position is clearly different from that of a structural analog of the tetraanionic form, 1,3,4,6-tetrathiapentalene-2,5-dione (TPD), observed at 1480 cm−1. To confirm the constancy of the formal charge of tto, variable temperature single-crystal X-ray diffraction measurements and resonance Raman spectroscopy of complex 1 were performed in 50 K increments from 300 K to 50 K. Upon cooling, the thermal movement of bulky TBA+ declines as seen in the thermal ellipsoid of carbon atoms in butyl groups (Fig. 1a and ESI†) and complex 1 is given space to distort its square-planar geometry around Ni atoms (Fig. 1b and Fig. S3, ESI†). Note that no drastic changes in CC bond length or energy of scattered light in Raman spectra were observed at any temperature measured, despite the changes in the overall molecular geometry as seen in Fig. S2 and S3 (ESI†). Therefore, the dianionic character of the tto moiety in complex 1 is stable over a wide temperature range. In addition, the thermogravimetric analysis demonstrated that complex 1 was thermally stable below 260 °C and the desorption temperature of the included DMSO was found to be 84 °C (Fig. S4, ESI†).
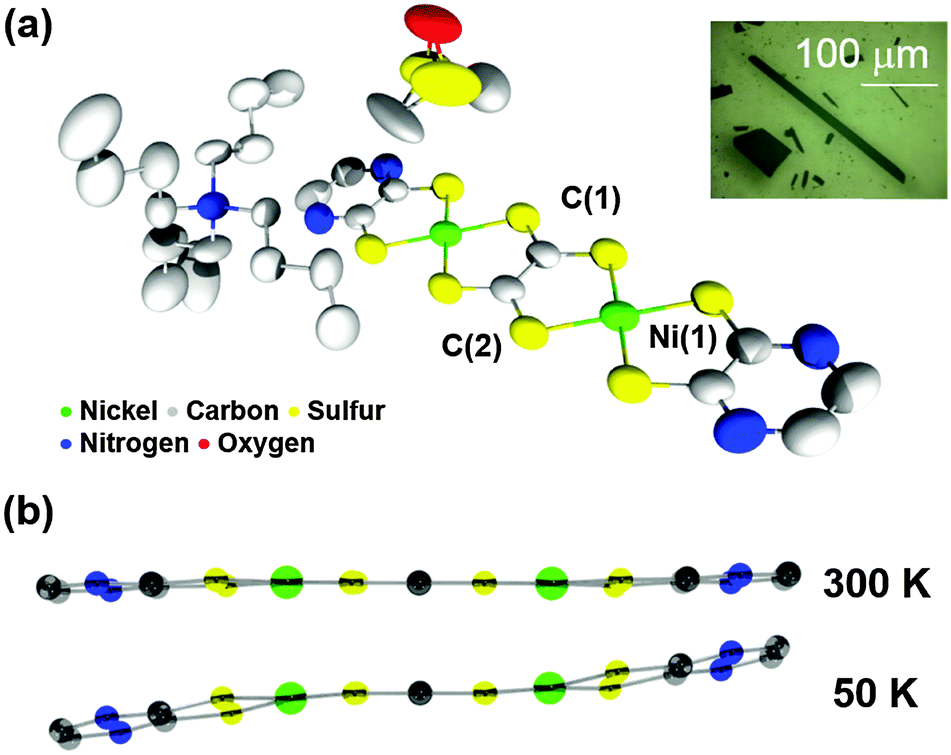 | ||
| Fig. 1 (a) The molecular structure of complex 1 in a single crystal at 300 K. Proton atoms are omitted for clarity. Inset shows crystals of complex 1. (b) Molecular geometries of complex 1 at 300 K and 50 K. | ||
To investigate the electronic structure of complex 1, its electrochemical behavior was monitored by cyclic voltammetry (Fig. 2a). The open circuit potential was found to be −0.6 eV, and two reversible reduction waves and one irreversible oxidation wave were observed. In the oxidation region, two large spike-shaped current responses were observed at Ea and Ec (0.21 eV and −0.90 eV vs. Fc/Fc+) which correspond to two-electron oxidation and re-reduction reactions of complex 1. Previous research indicates that these spike-shaped ones can be attributed to redissolution processes on the surface of the electrode that generate electrically conductive but insoluble forms of the oxidized complex 1.8 In addition, the large split between Ea and Ec (ΔE = 1.12 eV) may be attributed to structural changes in the oxidized state, for example a dimerization reaction.11 On the other hand, in the reduction region, two reversible redox reactions were clearly observed at −0.98 eV (E0′(1)) and −1.69 eV (E0′(2)) (vs. Fc/Fc+). These reduction processes were assigned to two successive one-electron redox reactions with a potential splitting of 0.7 eV caused by on-site coulomb repulsion. It is noteworthy that complex 1 has a narrow HOMO–LUMO energy gap (1.19 eV) that is the same as that of large porphyrin dimers with extended π-conjugated structures.4
 | ||
| Fig. 2 (a) Cyclic voltammograms of complex 1, TBA2[Ni(pdt)2] and 1,3,4,6-tetrathiapentalene-2,5-dione (TPD) in 0.1 M nBu4NClO4/DMSO solution at 100 mV s−1. Around −0.6 eV, the open triangle indicates the open circuit potential. (b) Calculated molecular orbitals. | ||
As shown in Fig. 2b, theoretical calculations suggest that the HOMO and LUMO of complex 1 are mainly located on two sites of Ni(pdt) (an antisymmetric combination of dxz and pz orbitals) and on the tto moiety (π*-orbital), respectively. It should be noted that the contribution of πsyn and πanti-orbitals on the tto moiety is especially prominent in the LUMO and LUMO+7 (Fig. S5a, ESI†). Therefore, the electronic structure of complex 1 is described as a D–A–D type. This consideration is theoretically and experimentally supported as follows. Firstly, as shown in Fig. 2a and Fig. S5b (ESI†), the electrochemically observed oxidation potential of TBA2[Ni(pdt)2] (−0.40 eV vs. Fc/Fc+) (considered to be D) and reduction potential of TPD (−0.26 eV vs. Fc/Fc+) considered as a two-electron reduced form of tto2− are close. This experimental result indicates that the energy levels of HOMO of [Ni(pdt)2]2− and LUMO of tto2− are close. Secondly, in terms of the donor–acceptor interaction, the dxz orbital of the Ni atom involved in calculated HOMO of [Ni(pdt)2]2− and pz orbitals of sulfur atoms involved in calculated LUMO of tto2− are in phase enough to interact with each other. These results indicate that complex 1 has a D–A–D -type of electronic structure, so that the empty π-orbital of the tto skeleton (b1u) strongly contributes to the reduction reaction accompanied by structural changes related to the orbital of b1u (Fig. 3).
 | ||
| Fig. 3 π-orbital of the tto skeleton and the expected redox behavior arising due to its noninnocent character. | ||
To examine the one-electron reduced state of complex 1, changes in the absorption spectrum were investigated using chemical and electrolytic reduction methods. As shown in Fig. 4, complex 1 has an intense absorption band at around 1150 nm assigned to the HOMO–LUMO transition by TD–DFT calculations. By adding CoCp2 as a reducing agent, the spectrum gradually changes, with an isosbestic point with the appearance of a red-shifted band at 1373 nm. Similar spectral transitions were also observed in the electrolytic absorption measurement by applying −0.9 V (vs. Fc/Fc+) (Fig. S6, ESI†). Electron spin resonance (ESR) measurements (Fig. 4, inset) revealed that the one-electron reduced state of complex 1 is an anionic radical with anisotropic g factors (g⊥ = 2.043 and g∥ = 2.009). Note that the LUMO of complex 1 was composed of not only π but also dxz orbitals because of the noninnocent nature of the tto skeleton, indicating that the reduced state of complex 1 is not a pure π-radical.12 These results strongly indicate that complex 1 can be defined as a compact D–A–D type of molecule, because the tiny tto skeleton can accept electrons. In addition, complex 1 also has an intense absorption in the NIR region in the solid state (Fig. S7, ESI†). Therefore, complex 1 can potentially be applied to molecular devices that can harvest NIR light, for example, thin-film solar cells. In these devices, complex 1 has the potential to exhibit a higher carrier mobility than other compounds because complex 1 with a small counter cation, such as alkali metals, is expected to be stacked closely and affords p- or n-type carriers in solid devices due to its compact and flat structure as well as rich redox properties based on the D–A–D nature.
 | ||
| Fig. 4 Absorption spectrum change induced by one-electron reduction of complex 1 in a DMSO solution using cobaltocene (CoCp2). ESR spectra in DMSO glass at 8 K (inset). | ||
In conclusion, a new compact, planar, D–A–D type of binickel complex with low-energy-gap characteristics was successfully synthesized. The variable temperature measurements of single-crystal X-ray diffraction and resonance Raman spectroscopy measurements demonstrated that the tto is in a stable dianonic state. Electrochemical analysis and theoretical calculations show that the electronic structure of complex 1 is of D–A–D type with a narrow HOMO–LUMO energy gap. Using electrolytic and chemical reduction, a strong absorption in the NIR region of complex 1 was confirmed by absorption spectroscopy, and assigned to the transition from HOMO to LUMO. Moreover, the one-electron reduced state of complex 1 has an anionic radical character that consists of not only π- but also d-orbitals because of the noninnocent nature of the tto moiety. The tto-bridged bimetal D–A–D system may be a promising molecule with compact, planar and low-energy-gap characteristics for molecular devices, especially for photoelectric conversion.
This work was partially supported by Core Research for Evolutional Science and Technology (CREST) Program No. 20350030 and No. 23245012 of Japan Science and Technology Agency (JST), and Grant-in-Aid for JSPS Fellows (No. 13J07305) from the Japan Society for the Promotion of Science (JSPS). Synchrotron XRD measurements were supported by the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal Numbers: 2013A1480, 2013B1400 and 2014A1406).
Notes and references
- (a) M. H. B. Stowell, T. M. McPhillips, D. C. Rees, S. M. Soltis, E. Abresch and G. Feher, Science, 1997, 276, 812–816 CrossRef CAS; (b) A. Cravino and N. S. Sariciftci, J. Mater. Chem., 2002, 12, 1931–1943 RSC; (c) A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed; (d) R. Rathore, S. V. Lindeman and J. K. Kochi, J. Am. Chem. Soc., 1997, 119, 9393–9404 CrossRef CAS; (e) P. Jonkheijm, N. Stutzmann, Z. Chen, D. M. de Leeuw, E. W. Meijer, A. P. H. J. Schenning and F. Würthner, J. Am. Chem. Soc., 2006, 128, 9535–9540 CrossRef CAS PubMed; (f) H. Alves, A. S. Molinari, H. Xie and A. F. Morpurgo, Nat. Mater., 2008, 7, 574–580 CrossRef CAS PubMed; (g) S. K. Park, S. Varghese, J. H. Kim, S.-J. Yoon, O. K. Kwon, B.-K. An, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2013, 135, 4757–4764 CrossRef CAS PubMed.
- (a) W.-S. Li, Y. Yamamoto, T. Fukushima, A. Saeki, S. Seki, S. Tagawa, H. Masunaga, S. Sasaki, M. Takata and T. Aida, J. Am. Chem. Soc., 2008, 130, 8886–8887 CrossRef CAS PubMed; (b) H. Choi, C. Baik, S. O. Kang, J. Ko, M.-S. Kang, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 327–330 CrossRef CAS PubMed; (c) A. B. Ricks, K. E. Brown, M. Wenninger, S. D. Karlen, Y. A. Berlin, D. T. Co and M. R. Wasielewski, J. Am. Chem. Soc., 2012, 134, 4581–4588 CrossRef CAS PubMed.
- (a) M. P. O'Neil, M. P. Niemczyk, W. A. Svec, D. Gosztola, G. L. Gaines and M. R. Wasielewski, Science, 1992, 257, 63–65 Search PubMed; (b) R. Y. Lai, E. F. Fabrizio, L. Lu, S. A. Jenekhe and A. J. Bard, J. Am. Chem. Soc., 2001, 123, 9112–9118 CrossRef CAS PubMed; (c) C.-H. Zhao, A. Wakamiya, Y. Inukai and S. Yamaguchi, J. Am. Chem. Soc., 2006, 128, 15934–15935 CrossRef CAS PubMed; (d) B. Breiten, Y.-L. Wu, P. D. Jarowski, J.-P. Gisselbrecht, C. Boudon, M. Griesser, C. Onitsch, G. Gescheidt, W. B. Schweizer, N. Langer, C. Lennartz and F. Diederich, Chem. Sci., 2011, 2, 88–93 RSC; (e) Z. Cai, Y. Guo, S. Yang, Q. Peng, H. Luo, Z. Liu, G. Zhang, Y. Liu and D. Zhang, Chem. Mater., 2013, 25, 471–478 CrossRef CAS.
- (a) A. Tsuda, H. Furuta and A. Osuka, Angew. Chem., Int. Ed., 2000, 39, 2549–2552 CrossRef CAS; (b) K. Susumu and M. J. Therien, J. Am. Chem. Soc., 2002, 124, 8550–8552 CrossRef CAS PubMed; (c) Y. Matsuo, M. Maruyama, S. S. Gayathri, T. Uchida, D. M. Guldi, H. Kishida, A. Nakamura and E. Nakamura, J. Am. Chem. Soc., 2009, 131, 12643–12649 CrossRef CAS PubMed.
- (a) F. Dumur, N. Gautier, N. Gallego-Planas, Y. Şahin, E. Levillain, N. Mercier, P. Hudhomme, M. Masino, A. Girlando, V. Lloveras, J. Vidal-Gancedo, J. Veciana and C. Rovira, J. Org. Chem., 2004, 69, 2164–2177 CrossRef CAS PubMed; (b) D. E. Janzen, M. W. Burand, P. C. Ewbank, T. M. Pappenfus, H. Higuchi, D. A. da Silva Filho, V. G. Young, J.-L. Brédas and K. R. Mann, J. Am. Chem. Soc., 2004, 126, 15295–15308 CrossRef CAS PubMed.
- (a) I. Janowska, F. Miomandre, G. Clavier, P. Audebert, J. Zakrzewski, K. H. Thi and I. Ledoux-Rak, J. Phys. Chem. A, 2006, 110, 12971–12975 CrossRef CAS PubMed; (b) R. Sakamoto, M. Murata and H. Nishihara, Angew. Chem., Int. Ed., 2006, 45, 4793–4795 CrossRef CAS PubMed; (c) R. Marczak, M. Wielopolski, S. S. Gayathri, D. M. Guldi, Y. Matsuo, K. Matsuo, K. Tahara and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 16207–16215 CrossRef CAS PubMed; (d) J. Ding, K. Feng, C.-H. Tung and L.-Z. Wu, J. Phys. Chem. C, 2011, 115, 833–839 CrossRef CAS.
- (a) M. D. Ward and J. A. McCleverty, J. Chem. Soc., Dalton Trans., 2002, 275–288 RSC; (b) M. Nomura, T. Cauchy, M. Geoffroy, P. Adkine and M. Fourmigué, Inorg. Chem., 2006, 45, 8194–8204 CrossRef CAS PubMed; (c) R. Sarangi, S. DeBeer George, D. J. Rudd, R. K. Szilagyi, X. Ribas, C. Rovira, M. Almeida, K. O. Hodgson, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 2316–2326 CrossRef CAS PubMed; (d) T. Kambe, R. Sakamoto, K. Hoshiko, K. Takada, M. Miyachi, J.-H. Ryu, S. Sasaki, J. Kim, K. Nakazato, M. Takata and H. Nishihara, J. Am. Chem. Soc., 2013, 135, 2462–2465 CrossRef CAS PubMed.
- (a) A. E. Pullen, R.-M. Olk, S. Zeltner, E. Hoyer, K. A. Abboud and J. R. Reynolds, Inorg. Chem., 1997, 36, 958–959 CrossRef CAS; (b) J.-F. Bai, J.-L. Zuo, Z. Shen, X.-Z. You, H.-K. Fun and K. Chinnakali, Inorg. Chem., 2000, 39, 1322–1324 CrossRef CAS; (c) A. S. Batsanov, M. R. Bryce, A. S. Dhindsa, J. A. K. Howard and A. E. Underhill, Polyhedron, 2001, 20, 537–540 CrossRef CAS; (d) K. Kubo, A. Nakao, H. M. Yamamoto and R. Kato, J. Am. Chem. Soc., 2006, 128, 12358–12359 CrossRef CAS PubMed.
- (a) R. Vicente, J. Ribas, S. Alvarez, A. Segui, X. Solans and M. Verdaguer, Inorg. Chem., 1987, 26, 4004–4009 CrossRef CAS; (b) Y. Sun, P. Sheng, C. Di, F. Jiao, W. Xu, D. Qiu and D. Zhu, Adv. Mater., 2012, 24, 932–937 CrossRef CAS PubMed.
- (a) C. Bianchini, C. Mealli, A. Meli, M. Sabat and P. Zanello, J. Am. Chem. Soc., 1987, 109, 185–198 CrossRef CAS; (b) H. A. Harris, D. R. Kanis and L. F. Dahl, J. Am. Chem. Soc., 1991, 113, 8602–8611 CrossRef CAS; (c) G. A. Holloway and T. B. Rauchfuss, Inorg. Chem., 1999, 38, 3018–3019 CrossRef CAS.
- W. E. Geiger, F. Barrière, R. J. LeSuer and S. Trupia, Inorg. Chem., 2001, 40, 2472–2473 CrossRef CAS.
- (a) M. M. R. Choudhuri, W. Kaim, B. Sarkara and R. J. Crutchley, Inorg. Chem., 2013, 52, 11060–11066 CrossRef CAS PubMed; (b) A. Mandal, H. Agarwala, R. Ray, S. Plebst, S. M. Mobin, J. L. Priego, R. Jiménez-Aparicio, W. Kaim and G. K. Lahiri, Inorg. Chem., 2014, 53, 6082–6093 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, 1H-NMR spectra, X-ray crystallographic information, TG analysis, temperature dependence of structural change obtained from the results of single crystal X-ray diffraction using a high intensity X-ray source in SPring-8, variable temperature measurements of resonance Raman spectroscopy, DFT calculations, absorption spectrum change obtained using electrolytic reduction and absorption spectrum diluted in KBr pellet. CCDC 1063807–1063813. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc06149k |
| This journal is © The Royal Society of Chemistry 2015 |