
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanism-based design of labile precursors for chromium(I) chemistry†
Eser S.
Akturk
,
Glenn P. A.
Yap
and
Klaus H.
Theopold
*
Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716, USA. E-mail: theopold@udel.educ
First published on 27th August 2015
Abstract
Dinitrogen complexes of the type TpR,RCr–N2–CrTpR,R are not the most labile precursors for Cr(I) chemistry, as they are sterically protected from obligatory associative ligand substitution. A mononuclear alkyne complex – TptBu,MeCr(η2-C2(SiMe3)2) – proved to be much more reactive.
Half a century after the discovery of the first dinitrogen complex, by Allen and Senoff,1 coordination compounds of the rather inert N2 molecule are still much sought after, due in large part to their substitutional lability and concomitant role as precursors for a wide variety of transition metal complexes.2 For example, our interest in the activation of O2 and other small molecules has benefited greatly from the availability of TptBu,MeCo(N2) and [(i-Pr2Ph)2nacnacCr]2(μ-η2:η2-N2), respectively.3,4 While these two molecules differ in the mode of coordination of the designated leaving group, both undergo facile ligand substitution to yield a plethora of compounds incorporating the TptBu,MeCo and (i-Pr2Ph)2nacnacCr fragments.5,6 We were interested in the intersection of these two chemistries, and accordingly we now report the preparation of dinitrogen complexes of various TpCr fragments, which exhibited some notable differences in reactivity.
KC8 reduction under nitrogen of blue TptBu,MeCr(THF)Cl in Et2O/THF (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at room temperature yielded green needles of [TptBu,MeCr]2(μ-η1:η1-N2) (1) in 42% yield (see ESI† for experimental detail and characterization of all compounds). The molecular structure of 1, as determined by X-ray diffraction, is shown in Fig. 1. The dinuclear complex contains a single N2 ligand bridging the two staggered TpCrI fragments, featuring end-on coordination of the dinitrogen to chromium. The N–N bond distance of 1.211(4) Å is substantially elongated over that of the free ligand (1.098 Å),7 and the Cr–N7 bond – at 1. 838(3) Å – is very short, certainly by comparison to the average Cr–NTp distance (2.198 Å). Both measures are consistent with strong π-backbonding from the low-valent chromium to the dinitrogen ligand. In accord with the crystallographically imposed inversion symmetry of 1, its IR spectrum (KBr) did not show a discernable N–N stretching vibration. 1 is a paramagnetic substance with isotropically shifted and broadened 1H NMR resonances. At room temperature, it has an effective magnetic moment of μeff = 3.9(1) μB, a possible interpretation of which is that the bridging N2 ligand mediates antiferromagnetic coupling between the two CrI (high-spin d5, S = 5/2) ions.
:1) at room temperature yielded green needles of [TptBu,MeCr]2(μ-η1:η1-N2) (1) in 42% yield (see ESI† for experimental detail and characterization of all compounds). The molecular structure of 1, as determined by X-ray diffraction, is shown in Fig. 1. The dinuclear complex contains a single N2 ligand bridging the two staggered TpCrI fragments, featuring end-on coordination of the dinitrogen to chromium. The N–N bond distance of 1.211(4) Å is substantially elongated over that of the free ligand (1.098 Å),7 and the Cr–N7 bond – at 1. 838(3) Å – is very short, certainly by comparison to the average Cr–NTp distance (2.198 Å). Both measures are consistent with strong π-backbonding from the low-valent chromium to the dinitrogen ligand. In accord with the crystallographically imposed inversion symmetry of 1, its IR spectrum (KBr) did not show a discernable N–N stretching vibration. 1 is a paramagnetic substance with isotropically shifted and broadened 1H NMR resonances. At room temperature, it has an effective magnetic moment of μeff = 3.9(1) μB, a possible interpretation of which is that the bridging N2 ligand mediates antiferromagnetic coupling between the two CrI (high-spin d5, S = 5/2) ions.
 | ||
| Fig. 1 The molecular structure of [TptBu,MeCr]2(μ-η1:η1-N2) (1, 30% probability level). Selected interatomic distances (Å) and angles (°): N7–N7A, 1.213(5); Cr–N7, 1.838(3), Cr–N1, 2.205(3); Cr–N3, 2.200(3); Cr–N5, 2. 190(3); NTp–Cr–NTp,avg, 87.3; NTp–Cr–N7avg, 127.2. | ||
With 1 in hand, we embarked on an exploration of its reactivity with a variety of small molecules. As expected, the low-valent dinitrogen complex reacted rapidly with molecules that yielded products in which the chromium was oxidized. Examples include O2, S8, N2O, and RN3. While the chalcogenide chemistry will be detailed elsewhere, we offer the product of the reaction of 1 with adamantyl azide, i.e. purple TptBu,MeCr![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NAd (2) as a representative example. 2 is the sole terminal imido complex of trivalent chromium.8 Its molecular structure is depicted in Fig. 2. The pseudo-tetrahedral complex features a linear imido ligand with a Cr–N distance of 1.687(2) Å; the latter is close the computationally predicted 1.708 Å for TptBu,MeCrNtBu.9 Consistent with the intermediate formal oxidation state of chromium it is also on the very long side of such distances.10 The effective magnetic moment of 2 measured μeff = 3.7(1) μB, which is consistent with a quartet spin ground state (d3, S = 3/2).
NAd (2) as a representative example. 2 is the sole terminal imido complex of trivalent chromium.8 Its molecular structure is depicted in Fig. 2. The pseudo-tetrahedral complex features a linear imido ligand with a Cr–N distance of 1.687(2) Å; the latter is close the computationally predicted 1.708 Å for TptBu,MeCrNtBu.9 Consistent with the intermediate formal oxidation state of chromium it is also on the very long side of such distances.10 The effective magnetic moment of 2 measured μeff = 3.7(1) μB, which is consistent with a quartet spin ground state (d3, S = 3/2).
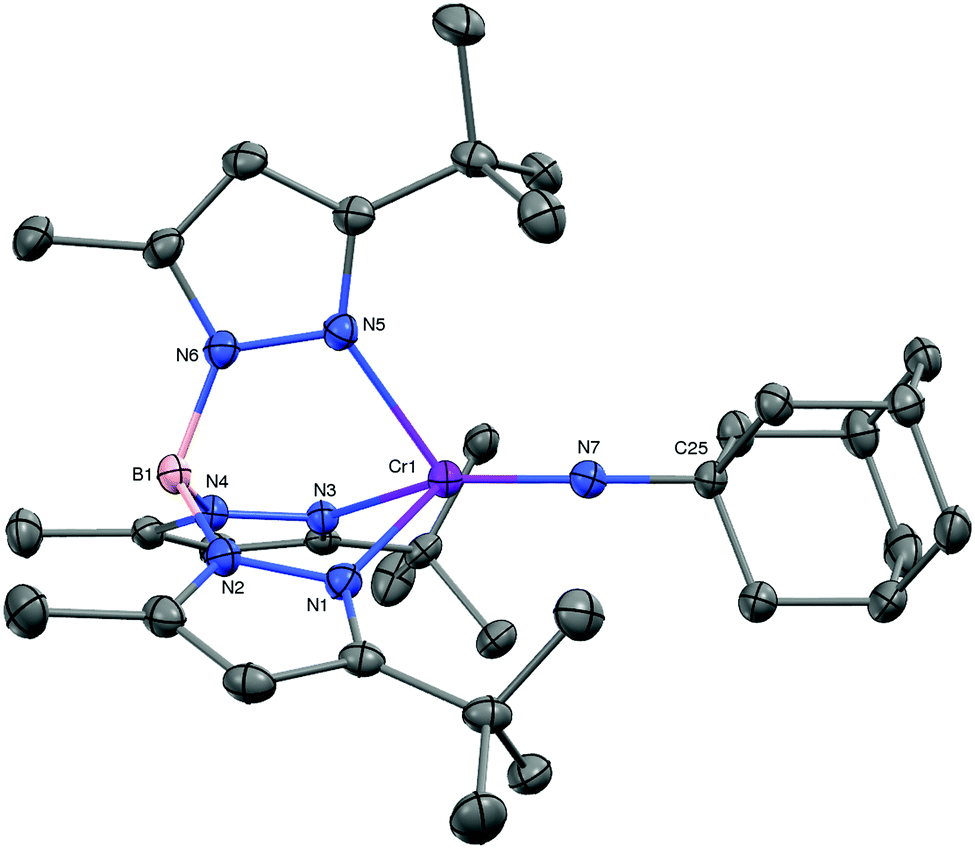 | ||
| Fig. 2 The molecular structure of TptBu,MeCrNAd (2, 30% probability level). Selected interatomic distances (Å) and angles (°): Cr–N7, 1.687(2); N7–C25, 1.455(3); Cr–N1, 2.132(2); Cr–N3, 2.151(2); Cr–N5, 2.160(2); Cr1–N7–C25, 178.8(2)°; NTp–Cr–NTp,avg, 88.0; NTp–Cr–N7avg, 126.7. | ||
To our surprise, reactions of 1 with good π-acceptors did either not proceed at all, or yielded decomposition products only after prolonged exposure. Thus, 1 did not react with alkenes (e.g., ethylene) or alkynes (e.g. 2-butyne), and lengthy exposure to an excess of CO (1 atm, 18 h) yielded only the ligand fragmentation product (tBu,MepzH)2Cr(CO)4, possibly due to traces of adventitious impurities (H2O?). We have reason to believe (vide infra) that all of these attempted reactions are thermodynamically favorable and would yield stable π-complexes. However, they apparently face insurmountable kinetic barriers, distinguishing 1 as a peculiarly substitution inert dinitrogen complex. To rationalize this disparity in reactivities, which has some precedent in titanium chemistry,11 we hypothesized that the reactions with oxidants may proceed via initial outer sphere electron transfer, thereby activating the Cr–N2 bond with respect to dissociation. Non-oxidizing ligands, on the other hand, may be forced to undergo an associative ligand substitution, because the Cr–N2 bond of 1 is too strong to permit a dissociative reaction path. The 13-electron configuration of the individual Cr atoms may make a ligand dissociation – yielding a bare, trigonal pyramidal 11-electron TptBu,MeCr fragment – energetically unfeasible. In this scenario, the effective steric shielding of the metal atoms by interleaving tert-butyl substituents of the opposing TptBu,Me ligands may prove impossible to penetrate, rendering the Cr–N2–Cr core of 1 impervious to ligand attack.
We then resolved to test the two essential pillars of this mechanistic hypothesis, namely (i) the lack of dissociation of 1, and (ii) the steric blocking of associative ligand substitution pathways. A dissociation of 1 in the absence of N2 must yield either one or two equivalents of TptBu,MeCr or a solvate thereof (TptBu,MeCr(S), S = Et2O, THF). Alternatively, in the presence of gaseous N2, an associative reaction with the latter may produce two equivalents of mononuclear intermediate TptBu,MeCr(N2). Either way, the reversible dissociation into mononuclear fragments should lead to scrambling of mixtures of suitably labeled dinuclear N2 complexes. In order to test this prediction we have prepared [TptBu,iPrCr]2(μ-N2) (3), a close analog of 1. 3 has been fully characterized, and selected structural parameters are listed in Table 1. In a control experiment, the reduction of an equimolar mixture of TptBu,MeCr(THF)Cl and TptBu,iPrCr(tBu,iPrpzH)Cl yielded a 1:2:1 mixture of 1, [TptBu,MeCr](μ-N2)[CrTptBu,iPr], and 3; the proportions of the products were measured by LIFDI-MS,12 which exhibited strong molecular ion (M+) peaks for these compounds. The ratio of the products did not change upon heating the mixture to reflux in THF. However, when a mixture of 1 and 3 in THF under vacuum was heated to 70 °C for two days, subsequent analysis of the mixture by LIFDI-MS showed no evidence for the formation of the mixed ligand complex ([TptBu,MeCr](μ-N2)[CrTptBu,iPr]). Similarly, when the same experiment was repeated under a N2 atmosphere, no signal for the mixed compound was detected in the mass spectrum. These results prove that 1 (and 3) do not detectably dissociate in THF solution, even when heated for prolonged periods. A dissociative mechanism (Id or D) for the ligand substitution of 1 is thereby ruled out.13
| Compound | 1 (TptBu,Me) | 3 (TptBu,iPr) | 4 (TpiPr,iPr) |
|---|---|---|---|
| N–N [Å] | 1.213(5) | 1.209(3) | 1.214(4) |
| Cr–N [Å] | 1.838(3) | 1.8395(16) | 1.773(2) |
| Cr–NTp [Å] | 2. 198 | 2.191 | 2.094 |
An alternative associative mechanism should be facilitated by lesser steric hindrance of the Tp ligands. To explore this possibility, we have prepared [TpiPr,iPrCr]2(μ-N2) (4). It is interesting to note that the N–N bond distance of 4 (see Table 1) does not significantly differ from those of 1 or 3; the extent of π-backbonding is apparently similar in all three compounds. However, the Cr–N distances in 4 are appreciably shorter (by 0.066(2) Å), suggesting that lesser steric interactions between the opposing ligands allow for a closer approach of the two TpCr fragments. Space filling models of 1 and 4 (see Fig. S3, ESI†) also suggest greater accessibility of the chromium centers in 4. In stark contrast to 1, exposure of 4 to 1 atm of CO(g) resulted in an immediate color change from violet to yellow and precipitation of octahedral TpiPr,iPrCr(CO)3 (5, see Fig. S4, ESI†). It appears that the diminished steric protection of Cr by the TpiPr,iPr-ligand causes a dramatic increase in the rate of ligand substitution; this observation argues strongly in favor of an associative substitution mechanism (Ia or A).
The results described above suggest that the preparation of coordination compounds of the TptBu,MeCrI fragment will require a precursor that is subject to facile associative ligand substitution; in all likelihood this will require a mononuclear structure to disrupt the molecular sheath protecting the Cr–N2–Cr core of 1. Based on related nacnacCr chemistry, and inspired by Rosenthal et al.,14 we selected TptBu,MeCr(η2-C2(SiMe3)2) (6) as a likely candidate.15 KC8 reduction of TptBu,MeCr(THF)Cl in Et2O/THF under vacuum in the presence of bis(trimethylsilyl)acetylene yielded brown crystals of 6 in 75% yield. The molecular structure of 6 (depicted in Fig. 3) features a severely distorted coordination environment, in which the centroid of the alkyne's triple bond is displaced from the B–Cr axis of the threefold symmetric TpCr fragment by 49°. This cis-divacant octahedral structure creates two symmetry equivalent openings for attack by external ligands. The relatively long Cr–Calkyne distances (2.048(2) and 2.084(2) Å) and the comparatively modest structural reorganization of the coordinated alkyne – by comparison with other complexes of the type TptBu,MeCr(η2-C2R2) (R = Me, Ph; see ESI†) – herald a rather tenuous hold of Cr upon this sterically encumbered alkyne. In accord with this notion, ‘spring-loaded’ 6 proved much more reactive toward ligand substitution than 1!
The reactions of 6 with various π-acceptors are summarized in Scheme 1; the molecular structures of the products – as determined by X-ray diffraction – are included in the ESI.† When carried out in ethereal solvents (THF, Et2O), these reactions were facile and proceeded in good yield. The carbonylation of 6 is notable in that it stopped short of the formation of TptBu,MeCr(CO)3 (i.e., the analog of 5). The actual product, κ2-TptBu,MeCr(CO)2(μ-η1:η1-CO)(Et2O)CrTptBu,Me (7) is best rationalized as the product of a disproportionation, resulting in a mixed-valent (Cr0CrII) isocarbonyl complex. The divalent chromium – formally a cation – has apparently lost its affinity for additional π-acids. The dinuclear ethylene complex, [κ2-TptBu,MeCr]2(μ-η2:η2-C2H4) (8), while a rare case of ethylene π-bonded to two metals,16 finds precedent in the analogous [(i-Pr2Ph)2nacnacCr]2(μ-η2:η2-C2H4).4 Like the latter, it did not react further with ethylene, exhibiting no activity for catalytic oligomerization or polymerization of ethylene.6a The irreversible reactions of 6 with less hindered alkynes were expected, being of interest mostly for the formation of pseudotetrahedral alkyne complexes 9 and 10, as evidenced by 1H NMR. More surprising was the observation that 6 reacted with N2 (1 atm), forming 1 and free alkyne quantitatively! The spontaneous substitution of an alkyne ligand by N2 is rather unusual. It is a measure of the instability and lability of 6 and – if additional proof was needed – suggests that it is an excellent precursor for TptBu,MeCrI chemistry.
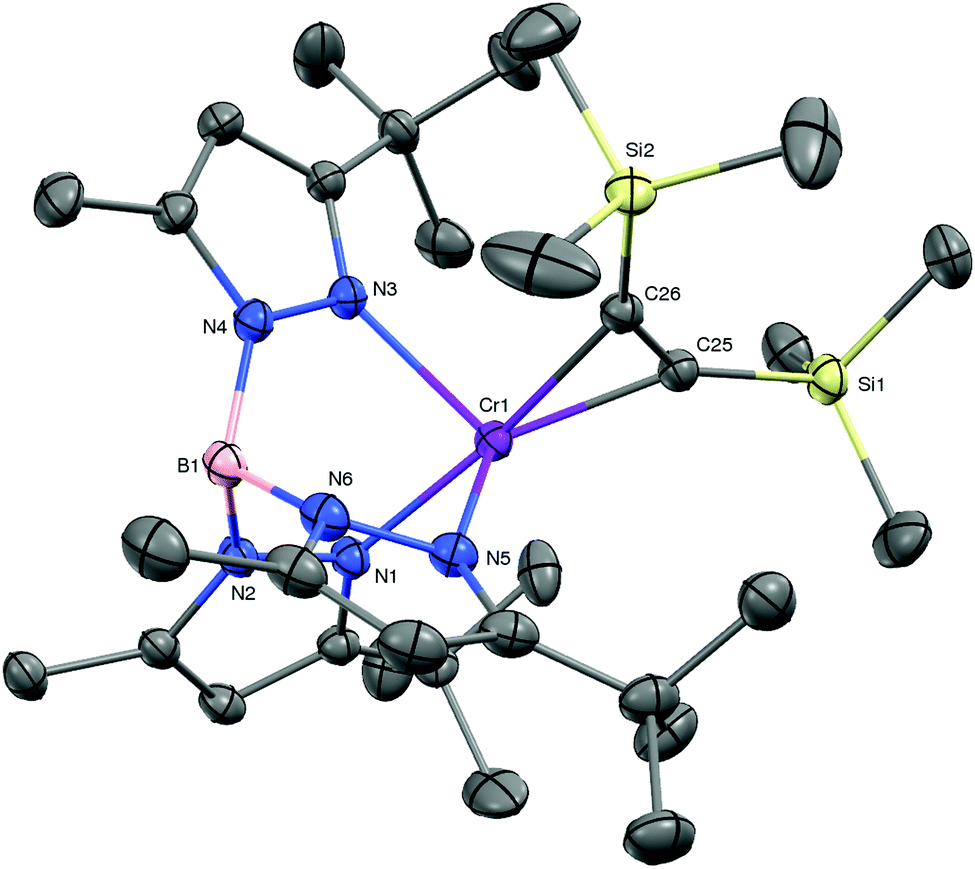 | ||
| Fig. 3 The molecular structure of TptBu,MeCr(η2-C2(SiMe3)2) (6, 30% probability level). Selected interatomic distances (Å) and angles (°): Cr–C25, 2.0480(19); Cr–C26, 2.0835(18); C25–C26, 1.288(3); Cr–N1, 2.1015(15); Cr–N3, 2.1614(16); Cr–N5, 2. 1504(16); NTp–Cr–NTp,avg, 87.7; N1–Cr–C25/C26centroid, 172.5; α (angle of deviation of alkyne centroid from B–Cr axis) = 49.3°. | ||
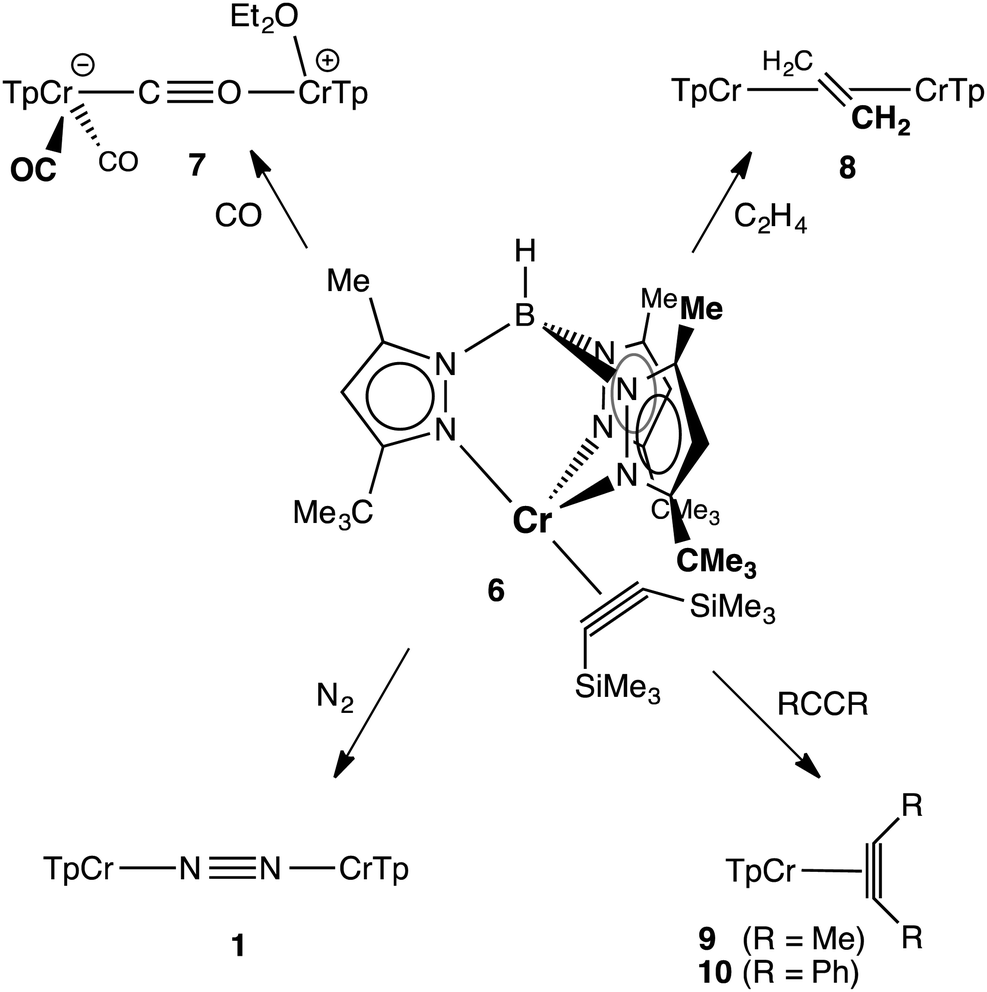 | ||
| Scheme 1 Ligand substitution reactions of 6. | ||
We are now exploring the small molecule activation chemistry of TpCr(I) fragments, judiciously using the synthons described above. The results of these studies will be reported in due course.
This research was supported by DOE (DE-FG02-92ER14273). Shared instrumentation for NMR, LIFDI-MS, and X-ray diffraction was supported by grants from NIGMS (1 P30 GM110758-01), NSF (CHE-1229234), and NSF (CRIF 1048367), respectively.
Notes and references
- A. D. Allen and C. V. Senoff, Chem. Commun., 1965, 621 RSC.
- (a) A. D. Allen, R. O. Harris, B. R. Loescher, J. R. Stevens and R. N. Whiteley, Chem. Rev., 1973, 73, 11 CrossRef CAS; (b) M. Hidai and Y. Mizobe, Chem. Rev., 1995, 95, 1115 CrossRef CAS; (c) T. A. Bazhenova and A. E. Shilov, Coord. Chem. Rev., 1995, 144, 69 CrossRef CAS; (d) M. P. Shaver and M. D. Fryzuk, Adv. Synth. Catal., 2003, 345, 1061 CrossRef CAS; (e) F. Tuczek and N. Lehnert, Angew. Chem., Int. Ed., 1998, 37, 2636 CrossRef CAS; (f) M. D. Fryzuk and S. A. Johnson, Coord. Chem. Rev., 2000, 200, 379 CrossRef; (g) B. A. MacKay and M. D. Fryzuk, Chem. Rev., 2004, 104, 385 CrossRef CAS; (h) S. Gambarotta and J. Scott, Angew. Chem., Int. Ed., 2004, 43, 5298 CrossRef CAS; (i) P. L. Holland, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004, p. 569 Search PubMed.
- J. W. Egan, B. S. Haggerty, A. L. Rheingold, S. C. Sendlinger and K. H. Theopold, J. Am. Chem. Soc., 1990, 112, 2445 CrossRef CAS.
- W. H. Monillas, G. P. Yap, L. A. MacAdams and K. H. Theopold, J. Am. Chem. Soc., 2007, 129, 8090 CrossRef CAS.
- (a) J. L. Detrich, R. Konečný, W. M. Vetter, D. Doren, A. L. Rheingold and K. H. Theopold, J. Am. Chem. Soc., 1996, 118, 1703 CrossRef CAS; (b) J. D. Jewson, L. M. Liable-Sands, G. P. A. Yap, A. L. Rheingold and K. H. Theopold, Organometallics, 1999, 18, 300 CrossRef CAS; (c) D. T. Shay, G. P. A. Yap, L. N. Zakharov, A. L. Rheingold and K. H. Theopold, Angew. Chem., Int. Ed., 2006, 45, 7870 CrossRef; (d) S. Thyagarajan, D. Shay, C. Incarvito, A. Rheingold and K. Theopold, J. Am. Chem. Soc., 2003, 125, 4440 CrossRef CAS.
- (a) W. H. Monillas, J. F. Young, G. P. A. Yap and K. H. Theopold, Dalton Trans., 2013, 42, 9198 RSC; (b) W. H. Monillas, G. P. A. Yap and K. H. Theopold, Inorg. Chim. Acta, 2011, 369, 103 CrossRef CAS.
- K. P. Huber and G. Herzberg, Constants of Diatomic Molecules, Van Nostrand Reinhold Co., New York, 1979 Search PubMed.
- A. A. Danopoulos, D. M. Hankin, G. Wilkinson, S. M. Cafferkey, T. K. N. Sweet and M. B. Hursthouse, Polyhedron, 1997, 16, 3879 CrossRef CAS.
- I. H. Wasbotten and A. Ghosh, Inorg. Chem., 2007, 46, 7890 CrossRef CAS.
- A search of CSD revealed 59 complexes with terminal imido ligands coordinated to Cr in oxidations states IV–VI (none lower). The mean Cr–N distance was 1.642(24) Å with a range of 1.550–1.692 Å.
- J. R. Hagadorn and J. Arnold, Inorg. Chem., 1997, 36, 2928 CrossRef CAS.
- H. B. Linden, Eur. J. Mass Spectrom., 2004, 10, 459 CrossRef CAS.
- (a) C. H. Langford and H. B. Gray, Ligand substitution processes, W.A. Benjamin, New York, 1965 Search PubMed; (b) R. B. Jordan, Reaction mechanisms of inorganic and organometallic systems, Oxford University Press, Oxford, New York, 2007 Search PubMed.
- (a) U. Rosenthal, V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg and V. B. Shur, Eur. J. Inorg. Chem., 2004, 4739 CrossRef CAS; (b) U. Rosenthal, V. V. Burlakov, P. Arndt, W. Baumann and A. Spannenberg, Organometallics, 2003, 22, 884 CrossRef CAS.
- F. Dai, G. P. A. Yap and K. H. Theopold, J. Am. Chem. Soc., 2013, 135, 16774 CrossRef CAS.
- (a) F. A. Cotton and P. A. Kibala, Inorg. Chem., 1990, 29, 3192 CrossRef CAS; (b) F. A. Cotton and P. A. Kibala, Polyhedron, 1987, 6, 645 CrossRef CAS; (c) F. A. Cotton, E. V. Dikarev, M. A. Petrukhina and R. E. Taylor, J. Am. Chem. Soc., 2001, 123, 5831 CrossRef CAS; (d) C. J. Burns and R. A. Andersen, J. Am. Chem. Soc., 1987, 109, 915 CrossRef CAS; (e) T. Takahashi, K. Kasai, N. Suzuki, K. Nakajima and E. Negishi, Organometallics, 1994, 13, 3413 CrossRef CAS; (f) R. Fischer, D. Walther, P. Gebhardt and H. Goerls, Organometallics, 2000, 19, 2532 CrossRef CAS; (g) T. Dube, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed., 1999, 38, 1432 CrossRef CAS; (h) P. J. Shapiro, W. D. Cotter, W. P. Schaefer, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 1994, 116, 4623 CrossRef CAS; (i) F. J. Fernandez, P. GomezSal, A. Manzanero, P. Royo, H. Jacobsen and H. Berke, Organometallics, 1997, 16, 1553 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and molecular structures of 3–5 and 7–10. CCDC 1058333–1058342. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc05993c |
| This journal is © The Royal Society of Chemistry 2015 |