
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A vanadium(IV) pyrazolate metal–organic polyhedron with permanent porosity and adsorption selectivity†
A. W.
Augustyniak
*a,
M.
Fandzloch
b,
M.
Domingo
c,
I.
Łakomska
b and
J. A. R.
Navarro
*c
aInstitute of Public Health and Environmental Protection, Batystowa 1B, 02-835 Warsaw, Poland. E-mail: adam.augustyniak1@gmail.com
bFaculty of Chemistry, Nicolaus Copernicus University, Gagarina 7, 87-100 Toruń, Poland
cDepartamento de Química Inorgánica, Universidad de Granada, Av. Fuentenueva S/N, 18071 Granada, Spain. E-mail: jarn@ugr.es
First published on 10th August 2015
Abstract
A vanadium(IV) pyrazolate-based open metal–organic polyhedron of [V3(μ3-O)O(OH)2(μ4-BPD)1.5(μ-HCOO)3] (BDP = benzene-1,4-bipyrazolate) formulation gives rise to a porous crystal structure exhibiting micro and mesoporosity which is useful for selective adsorption of gases.
Metal–organic polyhedra (MOPs) are discrete molecular architectures constructed via the coordination of metal ions and organic linkers to yield highly structured polyhedra. These systems usually possess a cavity able to incorporate host molecules thereby modifying their reactivity and dynamics.1 These features put these systems in close resemblance to biological self-assembled container supramolecules such as ferritin which play fundamental roles in regulating major physiological functions.2 MOPs are also interesting since they can be considered as the building units of MOFs,3 however, their discrete nature confers them a highly processable nature.4–6 On the other hand, vanadium ions are very attractive building units since their coordination diversity and redox activity give rise to a rich variety of predefined SBUs and catalytic activity.7–17 Noteworthily, the hard acidic nature of vanadium gives rise to the preferred use of O-donor linkers (i.e. phosphonate and carboxylates) for the construction of MOPs and MOFs.18–24 However, the enhanced basicity of some nitrogen-donor pyrazolate ligands has proven to give rise to highly stable constructs with Ni3(BTP)2 (BTP = 1,3,5-tris(1H-pyrazol-4-yl)benzene) being a remarkable example of high chemical and thermal stability making them potentially useful for application under harsh experimental conditions.25–31
In this communication, we show that the reaction of VCl3 and H2BDP (1,4-bi(1H-pyrazol-4-yl)benzene), in DMF under solvothermal conditions at 423 K, gives rise to single crystals of a system of [V3(μ3-O)O(OH)2(μ4-BPD)1.5(μ-HCOO)3] (1) formulation. The formation of formate anions should be regarded as a consequence of the hydrolysis of DMF solvent molecules during the solvothermal reaction. The crystallographic study of 1 reveals that its molecular structure is based on triangular trinuclear vanadium(IV) SBUs of the [V3(μ3-O)O(OH)2(μ-HCOO)3] type in which the vanadium centres exhibit a typically distorted octahedral VN2O4 stereochemistry.14 These triangular SBUs are connected by exotetradentate benzene-1,4-bipyrazolate (BDP) to give rise to the formation of tetrahedral MOPs with 1.2 nm inner cavities accessible through 0.85 nm wide triangular windows (see Fig. 1).‡ Noteworthily, MOP packing in the crystal structure follows a bcc arrangement which gives rise to 0.85 nm inter nanocage voids. Calculation of the Connolly surface of the crystal structure gives rise to high values of potentially accessible surface area for a probe molecule of 1.4 Å, namely 1660 m2 g−1 and an accessible pore volume of 0.130 cm3 cm−3 (Fig. 1C). In view of both the intra and inter cage porosity, we thought that this material might be useful for gas separation and purification purposes (see below). The physico-chemical nature of 1 was further studied by means of IR, EPR, TGA and XRPD (see the ESI†). Both room temperature EPR spectra of 1, exhibiting a sharp isotropic signal with a g value of 1.996, and IR spectra showing the apparition of the band at 953 cm−1 characteristic of V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O agree with the observed octahedral environment of vanadium(IV) centres. X-ray powder diffraction (XPRD) patterns of 1 are indicative of the phase purity of the bulk material. Thermogravimetric analysis of 1 in an atmosphere of air (Fig. S4, ESI†) reveals a significant weight loss (34%) of the material in the 373–503 K temperature range which corresponds to the loss of water and DMF solvent molecules. Above this temperature the desolvated material remains thermally stable up to 568 K. The permanent porosity of 1 was verified by the N2 adsorption isotherm at 77 K (Fig. 2), which reveals a type-I isotherm with a moderate value of BET surface area of 260 m2 g−1 and an adsorption capacity of 4.6 mmol g−1 at 0.8 P/Po. Additionally, the presence of a (type H4) hysteresis is indicative of textural mesoporosity arising from interparticle mesopores. Indeed, porosity distribution by density functional theory indicates, as implemented in Micromeritics software, the presence of both micropores of ca. 1.5 nm (inside cages) and mesopores of 5 nm (interparticle).
O agree with the observed octahedral environment of vanadium(IV) centres. X-ray powder diffraction (XPRD) patterns of 1 are indicative of the phase purity of the bulk material. Thermogravimetric analysis of 1 in an atmosphere of air (Fig. S4, ESI†) reveals a significant weight loss (34%) of the material in the 373–503 K temperature range which corresponds to the loss of water and DMF solvent molecules. Above this temperature the desolvated material remains thermally stable up to 568 K. The permanent porosity of 1 was verified by the N2 adsorption isotherm at 77 K (Fig. 2), which reveals a type-I isotherm with a moderate value of BET surface area of 260 m2 g−1 and an adsorption capacity of 4.6 mmol g−1 at 0.8 P/Po. Additionally, the presence of a (type H4) hysteresis is indicative of textural mesoporosity arising from interparticle mesopores. Indeed, porosity distribution by density functional theory indicates, as implemented in Micromeritics software, the presence of both micropores of ca. 1.5 nm (inside cages) and mesopores of 5 nm (interparticle).
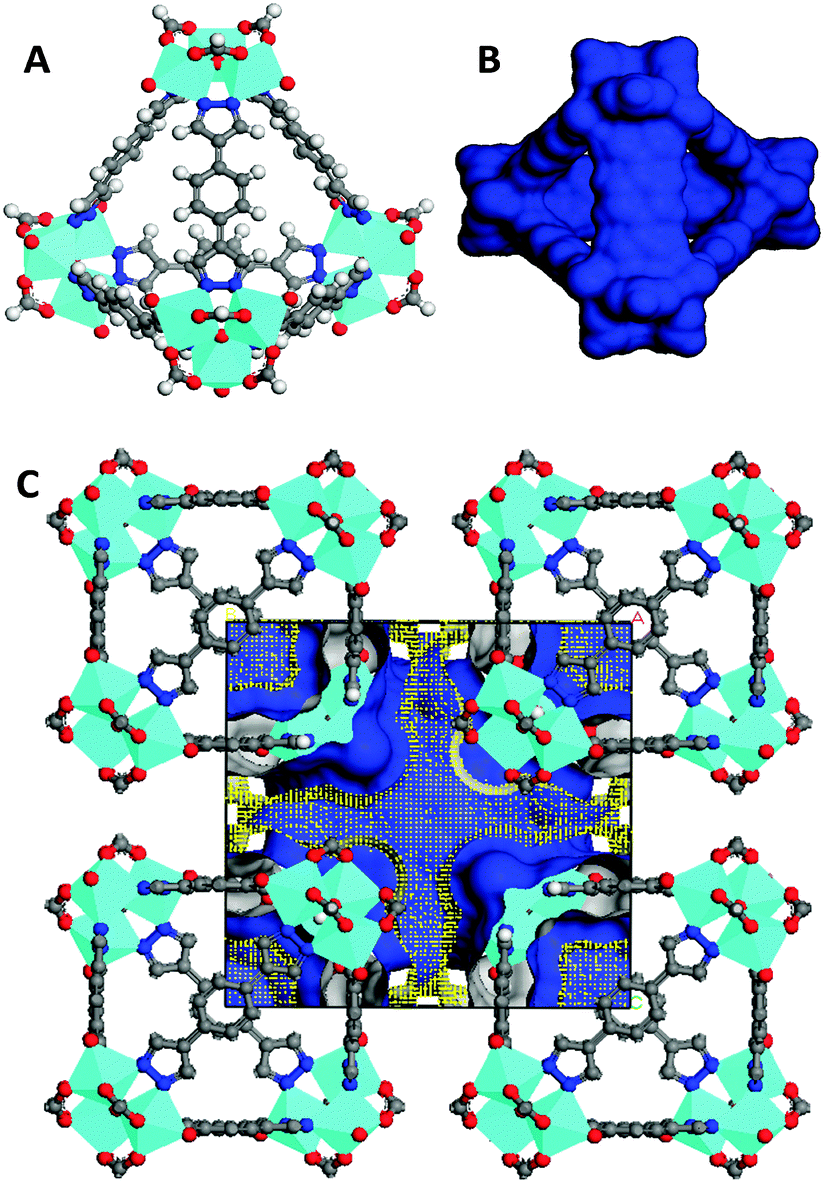 | ||
| Fig. 1 View of the molecular structure of a tetrahedral metal–organic polyhedron found in the crystal structure of 1 in the form of polyhedra (A), van der Waals surface (B) and Connolly surface of the crystal packing (C). | ||
 | ||
| Fig. 2 N2 adsorption (blue symbols) and desorption (red symbols) measured at 77 K. | ||
The appearance of mesoporosity, which is not observed in the crystal structure of 1 might be responsible for the observed higher pore volume accessibility than the calculated one from the crystal structure (the experimental accessible volume of 0.150 cm3 cm−3vs. the calculated value of 0.130 cm3 cm−3), should be related to a partial loss of supramolecular ordering after sample desolvation. By contrast, the observed BET surface area is considerably lower than the calculated one from the Connolly surface for a probe molecule of 1.4 Å (260 m2 g−1vs. 1660 m2 g−1) which might be indicative of poor accessibility of the probe molecule to the whole crystal porosity after sample activation (see below).
Indeed, XRPD of 1 after thermal activation indicates that although the original XRPD pattern is maintained a certain degree of amorphization is obvious which may give rise to the observed hierarchical pore structure and to the observed slow diffusion of N2 molecules inside the pore structure which is manifested by long equilibration times during the adsorption measurements.
Once we proved the accessible nature of the porous structure of 1 we proceed to evaluate its possible utility in gas and vapor separation processes, namely CO2 capture from flue gas and benzene/cyclohexane separation. Fig. 3 shows the CO2 adsorption isotherms of 1 at 273 and 298 K as well as a comparison of N2 and CO2 adsorption isotherms at 273 K. The CO2 adsorption isotherms of 1 follow a type-I behavior in the studied pressure range. The isosteric heats of CO2 adsorption were evaluated according to the Clausius–Clapeyron (eqn (1)) from the adsorption isotherms measured at 273 and 298 K:
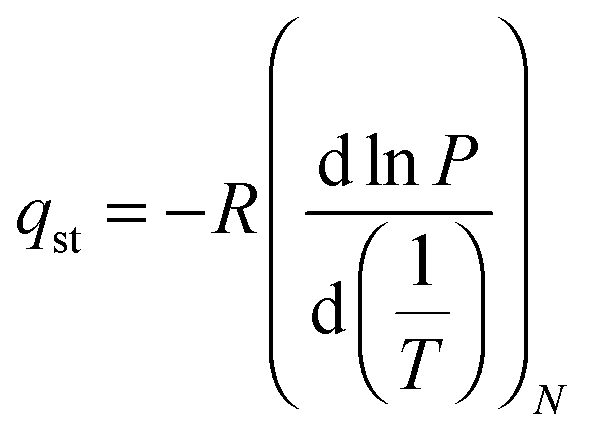 | (1) |
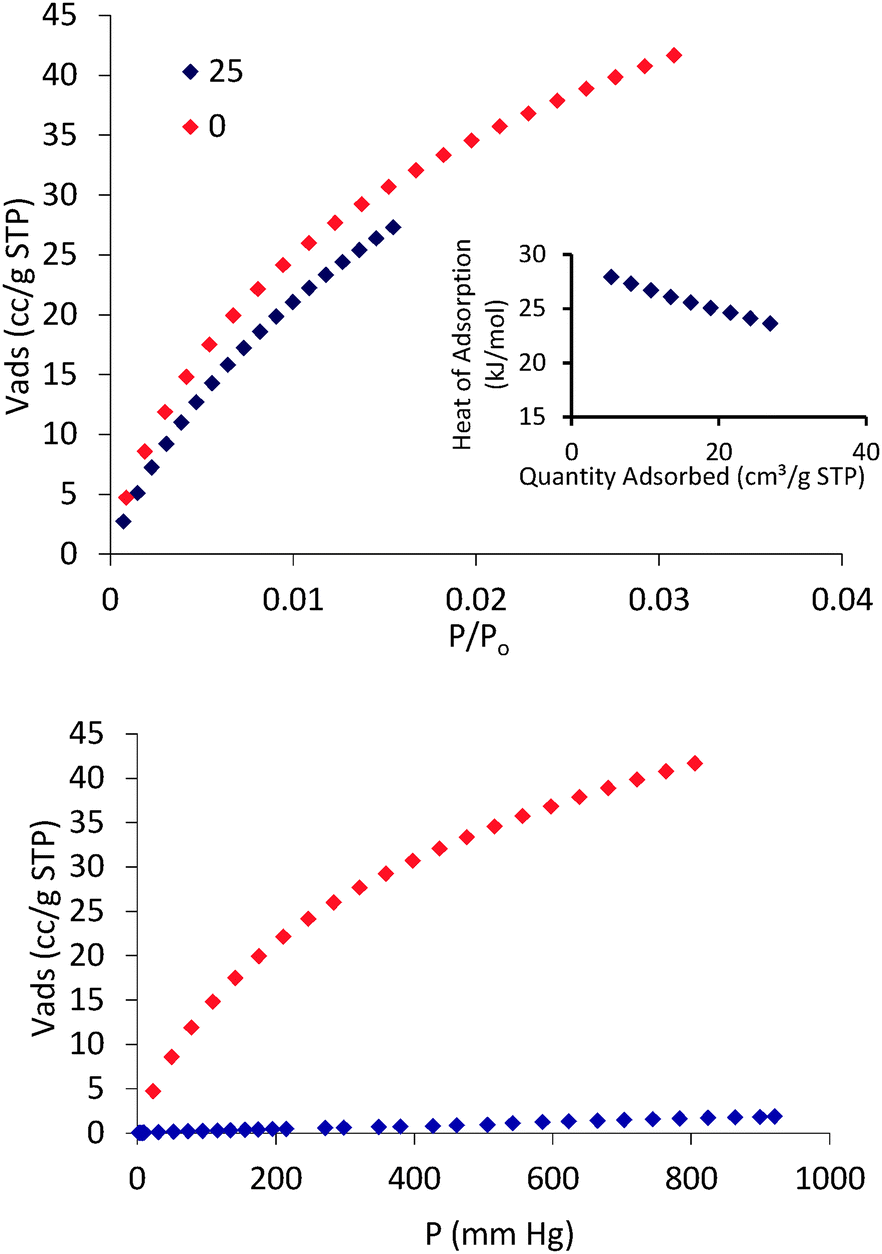 | ||
| Fig. 3 CO2 adsorption isotherms measured at 273 K (red) and 298 K (blue) (top), CO2 isosteric heat of adsorption (inset in top) and CO2/N2 selectivity at 273 K (down). | ||
In order to prove that the adsorption selectivity of 1 towards CO2 is maintained towards gas mixtures we have prepared a chromatographic column of this material (0.35 g) packed in a 15 cm long glass column with a 5 mm inner diameter and tested the separation process towards a simulated flue gas (86% of N2 and 14% CO2). For this purpose, we proceeded to activate the packed chromatographic column under an He flow (10 mL min−1) at 403 K and after cooling down to 273 K we flowed a gas mixture containing 1.4 mL min−1 of CO2 and 8.6 mL min−1 of N2. The results show (Fig. 4) that CO2 is selectively retained for 160 s which corresponds to the adsorption capacity of 0.5 mmol of CO2 g−1 while N2 does not interact with the adsorbent.
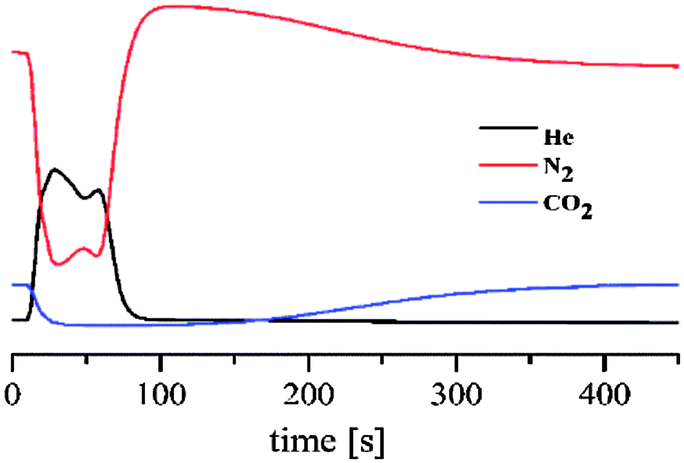 | ||
| Fig. 4 N2/CO2 separation breakthrough experiments at 273 K of a column packed MOP 1. | ||
In order to further prove the adsorption selective properties of this system we have also essayed the possible suitability of this material for the shape selective separation of benzene (b.p. 80.1 °C) and cyclohexane (b.p. 80.7 °C) since separation by distillation is not suitable since the b.p. of both compounds differ in less than 1 K.31 With this aim we have carried out variable-temperature pulse gas chromatography experiments in the 383–423 K temperature range with a complex gas mixture (methane, cyclohexene and benzene) in order to find out the possible resolution of this gas mixture by 1 (Fig. 5). The results show that 1 gives rise to negligible interactions with methane in the studied temperature range which is useful as a reference.
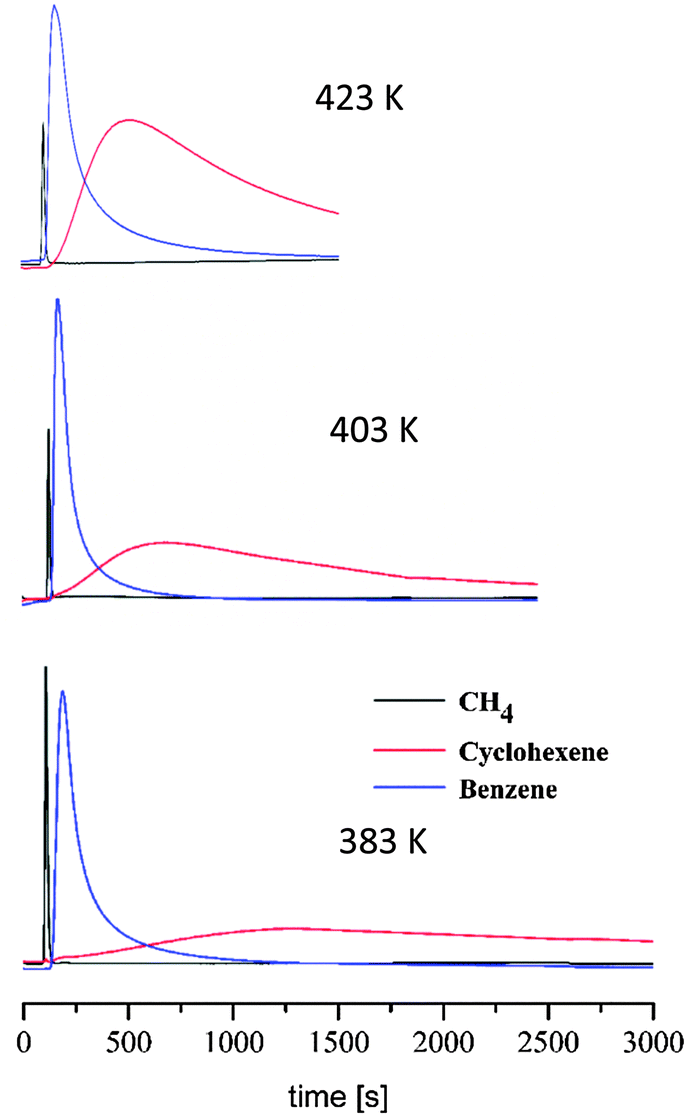 | ||
| Fig. 5 Variable-temperature pulse gas chromatography experiments of an equimolecular CH4/benzene/cyclohexene gas mixture passed through a chromatographic column packed with vanadium nanocages using a He flow of 10 mL min−1. | ||
Noteworthily, while the benzene interaction seems to be strong as manifested by long retention times, broad peaks and high adsorption heat value (−28.3 kJ mol−1), the interaction with cyclohexene appears to be much weaker as manifested by lower retention times, sharper chromatographic peaks and low adsorption heat value (−2.0 kJ mol−1). Indeed, αbenzene/cyclohexane selectivity values of 10 are found which should be attributed to the smaller size of benzene compared to cyclohexane which may facilitate the diffusion of benzene inside the MOP cavities justifying its higher retention while cyclohexane molecules are probably interacting with the external surface of the MOP microcrystalline powder.
In summary, a new vanadium MOP has been synthesized and fully characterized. It should be noted that in spite of the discrete nature of 1 MOP, the crystallographic study as well as the static and dynamic adsorption experiments reveal the accessibility of both the intra and intercage space to gas and VOC molecules giving rise to selective adsorption processes which are useful for carbon capture applications as well as shape separation of challenging benzene/cyclohexane mixtures.
The authors thank Dr A. Lewińska of Wrocław University (EPR interpretation). We also thank Dr A. Rodriguez-Dieguez and Dr L. M. Rodriguez-Albelo of University of Granada for single crystal measurement and Connolly surface calculation. JARN is grateful to the Spanish Ministry of Economy and Competitiveness (CTQ2014-53486-R) for generous funding.
Notes and references
- H. Vardhana and F. Verpoort, Adv. Synth. Catal., 2015, 357, 1351 CrossRef PubMed.
- F. Carmona, O. Palacios, N. Gálvez, R. Cuesta, S. Atrian, M. Capdevila and J. M. Domínguez-Vera, Coord. Chem. Rev., 2013, 257, 2752 CrossRef CAS PubMed.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141 RSC.
- Y. Han, J.-R. Li, Y. Xie and G. Guo, Chem. Soc. Rev., 2014, 43, 5952 RSC.
- L.-B. Sun, J.-R. Li, W. Lu, Z.-Y. Gu, Z. Luo and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 15923 CrossRef CAS PubMed.
- H. Vardhana and F. Verpoort, Adv. Synth. Catal., 2015, 357, 1351 CrossRef PubMed.
- I. Swart, A. Fielicke, B. Redlich, G. Meijer, B. M. Weckhuysen and F. M. F. de Groot, J. Am. Chem. Soc., 2007, 129, 2516 CrossRef CAS PubMed.
- C. Ratsch, J. Chem. Phys., 2005, 122, 124302 CrossRef CAS PubMed.
- I. S. Tidmarsh, Inorg. Chem., 2007, 46, 9743 CrossRef CAS PubMed.
- A. Butler and C. J. Carrano, Coord. Chem. Rev., 1991, 109, 61 CrossRef CAS.
- Z. Janas, D. Wiśniewska, L. B. Jerzykiewicz, P. Sobota, K. Drabent and K. Szczegot, Dalton Trans., 2007, 2065 RSC.
- Z. Janas and P. Sobota, Coord. Chem. Rev., 2005, 249, 2144 CrossRef CAS PubMed.
- C. Lorber, F. Wolff, R. Choukroun and L. Vendier, Eur. J. Inorg. Chem., 2005, 2850 CrossRef CAS PubMed.
- F. Wolff, C. Lorber, R. Choukroun and B. Donnadieu, Inorg. Chem., 2003, 42, 7839 CrossRef CAS PubMed.
- E. Y. Tshuva, N. Gendeziuk and M. Kol, Tetrahedron Lett., 2001, 42, 6405 CrossRef CAS.
- M. Sutradhar, G. Mukherjee, M. G. B. Drew and S. Ghosh, Inorg. Chem., 2006, 45, 5150 CrossRef CAS PubMed.
- D. Rehder, Bioinorganic Vanadium Chemistry, John Wiley & Sons, Ltd., New York, 2008 Search PubMed.
- V. Chandrasekhar, A. Dey, T. Senapatia and E. C. Sanudo, Dalton Trans., 2012, 41, 799 RSC.
- K. Barthelet, J. Marrot, D. Riou and G. Ferey, Angew. Chem., Int. Ed. Engl., 2002, 41, 281 CrossRef CAS.
- K. Barthelet, D. Riou, M. Nogues and G. Ferey, Inorg. Chem., 2003, 42, 1739 CrossRef CAS PubMed.
- I. J. Kang, N. A. Khan, E. Haque and S. H. Jhung, Chemistry, 2011, 17, 6437 CrossRef CAS PubMed.
- W. Kaveevivitchai, X. Wang, L. Liu and A. J. Jacobson, Inorg. Chem., 2015, 54, 1822 CrossRef CAS PubMed.
- Y. Y. Liu, S. Couck, M. Vandichel, M. Grzywa, K. Leus, S. Biswas, D. Volkmer, J. Gascon, F. Kapteijn, J. F. M. Denayer, M. Waroquier, V. Van Speybroeck and P. Van Der Voort, Inorg. Chem., 2013, 52, 113 CrossRef CAS PubMed.
- A. Phan, A. U. Czaja, F. Gandara, C. B. Knobler and O. M. Yaghi, Inorg. Chem., 2011, 50, 7388 CrossRef CAS PubMed.
- C. Montoro, F. Linares, E. Quartapelle Procopio, I. Senkovska, S. Kaskel, S. Galli, N. Masciocchi, E. Barea and J. A. R. Navarro, J. Am. Chem. Soc., 2011, 133, 11888 CrossRef CAS PubMed.
- N. Masciocchi, S. Galli, A. Sironi, E. Cariati, M. A. Galindo, E. Barea, M. A. Romero, J. M. Salas, J. A. R. Navarro and F. Santoyo-González, Inorg. Chem., 2006, 45, 7612 CrossRef CAS PubMed.
- N. Masciocchi, S. Galli, G. Tagliabue, A. Sironi, O. Castillo, A. Luque, G. Beobide, W. Wang, M. A. Romero, E. Barea and J. A. R. Navarro, Inorg. Chem., 2009, 48, 3087 CrossRef CAS PubMed.
- E. Barea, C. Montoro and J. A. R. Navarro, Chem. Soc. Rev., 2014, 43, 5419 RSC.
- S. Galli, N. Masciocchi, V. Colombo, A. Maspero, G. Palmisano, F. J. López-Garzón, M. Domingo-García, I. Fernández-Morales, E. Barea and J. A. R. Navarro, Chem. Mater., 2010, 22, 1664 CrossRef CAS.
- V. Colombo, C. Montoro, A. Maspero, N. Masciocchi, S. Galli, E. Barea and J. A. R. Navarro, J. Am. Chem. Soc., 2012, 134, 12830 CrossRef CAS PubMed.
- E. Quartapelle-Procopio, F. Linares, C. Montoro, V. Colombo, A. Maspero, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2010, 49, 7308 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of MOP and linker molecules, XPRD patterns, IR, TG and EPR measurements. CCDC 1412183. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc05913e |
‡ Crystal data for 1: C84H68N24O40V12, M = 2664.87 g mol−1, cubic, space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m, a = b = c = 20.084(5) Å, V = 8101(3) Å3, Z = 2, ρcalc = 1.084 mg m−3, T = 100 K, λMoKα = 0.71073 Å, R(F, F2 > 2σ) = 0.0683, Rw(F2, all data) = 0.1648 for 497 unique reflections, goodness-of-fit = 1.250. CCDC 1412183. 3m, a = b = c = 20.084(5) Å, V = 8101(3) Å3, Z = 2, ρcalc = 1.084 mg m−3, T = 100 K, λMoKα = 0.71073 Å, R(F, F2 > 2σ) = 0.0683, Rw(F2, all data) = 0.1648 for 497 unique reflections, goodness-of-fit = 1.250. CCDC 1412183. |
| This journal is © The Royal Society of Chemistry 2015 |