
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomimetic vaterite formation at surfaces structurally templated by oligo(glutamic acid) peptides†
Hao
Lu
a,
Matthew A.
Hood‡
a,
Sergio
Mauri
a,
Joe E.
Baio
b,
Mischa
Bonn
a,
Rafael
Muñoz-Espí
a and
Tobias
Weidner
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, D-55128 Mainz, Germany. E-mail: weidner@mpip-mainz.mpg.de; Tel: +49 (0) 6131 379 547
bSchool of Chemical, Biological, and Environmental Engineering, Oregon State University, OR, USA
First published on 10th September 2015
Abstract
Previous studies have reported that the metastable vaterite phase of calcium carbonate can be stabilized in solution by acidic additives. Here we demonstrate that vaterite can also be stabilized directly at surfaces by engineered peptides. Our data show that the mineralisation occurs in a ‘self-templating’ process where calcium ions restructure the peptide backbone, which in turn allows for effective vaterite precipitation.
In nature, biomacromolecules, like peptides, proteins and polysaccharides, are frequently employed to control the formation of minerals, such as calcium carbonate (CaCO3), which can be found in pearl, nacre, and other invertebrate hard tissue.1–9 To better understand this biomineralisation process, previous work has involved extracting peptides or proteins from mollusk shells and other invertebrates, and determining whether these biomacromolecules function as organic templates to regulate the crystallization of calcium carbonate.1,3–5 Typically, CaCO3 crystallizes into three anhydrous polymorphs: calcite, aragonite, and vaterite. Thermodynamically, vaterite is the least stable phase and accordingly it occurs rarely as a geological mineral in sediments.10 At the same time, for material science, vaterite is a highly attractive CaCO3 polymorph with potential applications in implant design, regenerative medicine, bone implants, targeted drugs, and personal care products.11–15 The observation that formation of vaterite can be triggered in vitro by soluble biomimetic molecules11,12,16–19 has prompted numerous fundamental studies on the mechanisms of its nucleation, growth, and stabilization, aiming to improve understanding of the biomineralisation process of CaCO3.16,17
Previous studies in solution have shown that vaterite can be stabilized in the presence of (bio-)molecules such as polymers,11,12 amino acids,16,19 and peptides.16–18 It is believed that the acidic residues of these molecules act as a molecular modulator: they display strong, preferential binding to specific mineral phases such as calcite in calcium carbonate and thus favour vaterite formation.10,16,18,20 The most representative examples in this context are oligo(glutamic acid) peptides: at basic pH deprotonated acidic residues exhibit greater affinity towards specific calcite crystal planes, inhibit growth along the respective crystal directions and thereby promote vaterite growth.16,17 The above process can occur in bulk surroundings, or in particular near the surface: previously, Fischer et al. have found that carboxylic-functionalized latex polymer particles can be used to control the crystallization of a similar calcium-based mineral (calcium oxalate), and moreover, these modified particles are quite densely deposited onto the controlled growing crystal plane.21 Based on these results, we hypothesize the impact of acidic groups on vaterite formation is even stronger near surfaces rather than in solution.
In this communication we demonstrate that vaterite can also be stabilized on inorganic surfaces by peptides designed to carry acidic side chains. Direct surface precipitation can be advantageous for the design of biocompatible implant surfaces and targeted drug particles. To glean information about the surface mineralisation process we study the interaction between surface-bound peptides and CaCO3 minerals.
As a model system, we used a thiol-terminated oligo(glutamic acid) peptide (Glu5, see Scheme 1) attached to gold surface. Upon interaction with the calcium precursors, the chemisorbed peptide monolayer acted as a structural template, mediating the precursors so that vaterite was preferentially expressed. The vaterite polymorph structure obtained was characterized by X-ray diffraction (XRD) and scanning electron microscopy (SEM) measurements. In addition, the structural changes in the peptide template accompanying the vaterite formation were monitored by a combination of near edge X-ray absorption fine structure (NEXAFS) and sum frequency generation (SFG) spectroscopy.
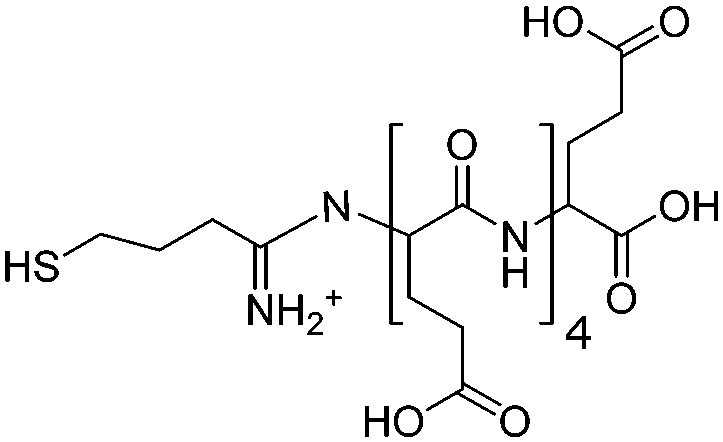 | ||
| Scheme 1 Structure of thiol-terminated oligo(glutamic acid) peptide, abbreviated as Glu5. | ||
The formation of a contamination-free and densely packed Glu5 peptide monolayer on Au was confirmed by X-ray photoelectron spectroscopy (XPS). The derived atomic composition averaged over three different samples of Glu5 films is provided in the ESI† (Table S1). The data clearly show the expected presence of carbon, nitrogen, and oxygen, and the results are in general agreement with the theoretical composition of Glu5 molecules preserving full chemical integrity. N 1s and S 2p spectra are shown in Fig. 1(a) and (b), respectively. A dodecanethiol (DDT) self-assembled monolayer (SAM) was included as reference with well documented surface structure.22 The N 1s spectrum shown in Fig. 1(a) exhibits a single emission near 400 eV related to the amide nitrogen atoms within the Glu5 backbone. The nitrogen signal is absent in the DDT control spectrum. The chemical attachment of Glu5 on the gold surface was determined using S 2p spectra, as shown in Fig. 1(b). The spectra exhibit a S 2p3/2,1/2 doublet at a binding energy (BE) of 161.9 to 162.0 eV, which can be assigned to thiolate-type sulfur bonded directly to the Au surface, while no traces of physisorbed or oxidized species were observed in the spectra. This BE value also agrees well with the analogous values previously observed for aliphatic and aromatic self-assembled monolayers (SAMs) on gold substrates.23,24 In addition to the analysis of the peak energies in the spectra, the packing density of the Glu5 peptide SAM was estimated from the intensity ratios of the S 2p and Au 4f emissions, following the approach of ref. 25 and 26. As a reference system, the DDT SAM with well-known packing density of 4.62 × 1014 molecules per cm2 was used.23,24 The derived packing density is 3.58 × 1014 molecules per cm2, which is in the range for common SAMs,23,26 but much higher than that for larger peptides or protein on the surface, showing that the packing is very dense.27 By comparison, the packing density for 15-amino acid peptides is ∼0.25 × 1014 molecules per cm2.28 Here the dense Glu5 packing ensures a high local density of acidic residues at the surface and, possibly, provides improved bonding opportunity for crystallizing CaCO3.
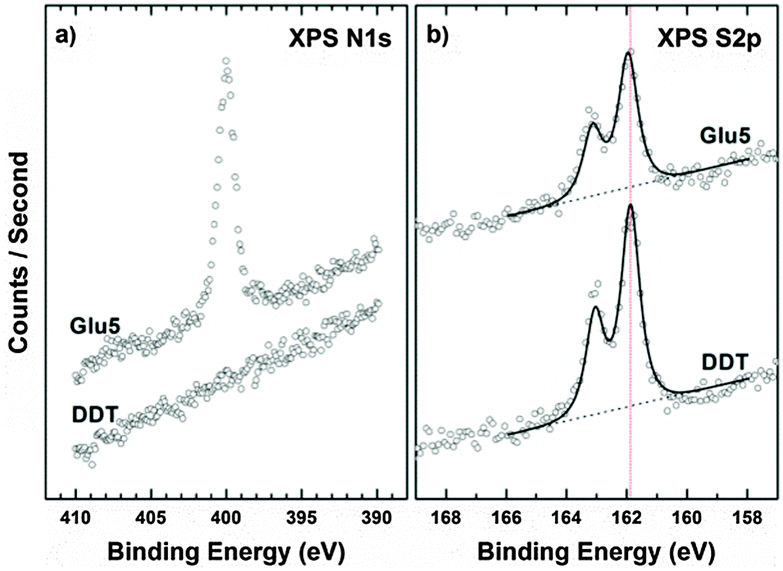 | ||
| Fig. 1 N 1s (a) and S 2p (b) XPS spectra of Glu5 peptide and DDT monolayers on Au substrate. The S 2p spectra in (b) are both fitted with one doublet, and the red dashed line highlights the binding energy position of the emission. | ||
The mineralisation of calcium carbonate on the functionalized gold surfaces was studied by XRD and the resulting patterns are shown in Fig. 2. The main reflection at 2θ = 32.8°, which can be unambiguously assigned to vaterite, demonstrates the stabilization of this crystal modification on Glu5 peptide layer, while calcite is the only phase obtained on the DDT-coated surface. The stabilization of vaterite on Glu5 SAM is further supported by the SEM images, shown in Fig. 3. The typical spherical vaterite polymorph can be clearly seen on the Glu5 SAM, while the DDT coated surface results in typical rhombohedral calcite structure. Here the obtained vaterite structure survived after repeated rinsing with water for salt removal, and was also stable under ambient condition for months.
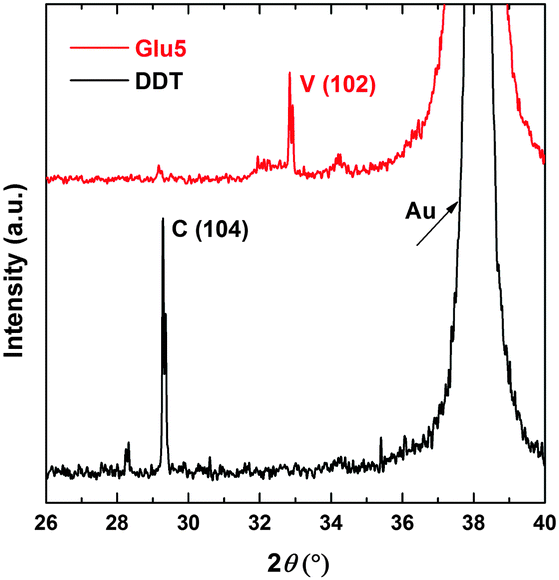 | ||
| Fig. 2 XRD pattern of calcium carbonate formed on Glu5 peptide (red) and DDT (black) monolayers on Au substrate. (V) Vaterite; (C) Calcite. | ||
 | ||
| Fig. 3 SEM images of calcium carbonate polymorphs formed on DDT: calcite (a and b) and Glu5 peptide monolayers: vaterite (c and d) on Au. | ||
To obtain information about the mechanism by which the Glu5 monolayer induces vaterite formation, we applied NEXAFS spectroscopy to investigate structural changes within the Glu5 SAM when interacting with the mineral precursors. NEXAFS spectra exhibit characteristic resonances related to electronic transitions from atomic core levels to unoccupied molecular orbitals making this technique very sensitive to molecular bonds and the local environment around the probed element.29,30 For protein and peptides on surfaces, NEXAFS can provide important information about their structure by probing the conformation of the backbone amide bonds at the nitrogen K-edge.28,31Fig. 4 (left) shows nitrogen NEXAFS spectra of Glu5 SAMs before and after vaterite formation. As expected, the spectra are dominated by a broad σ* resonance at ∼406 eV attributed to N–C and N–H orbitals, and a π* transition at ∼401 eV assigned to backbone amide bonds.31
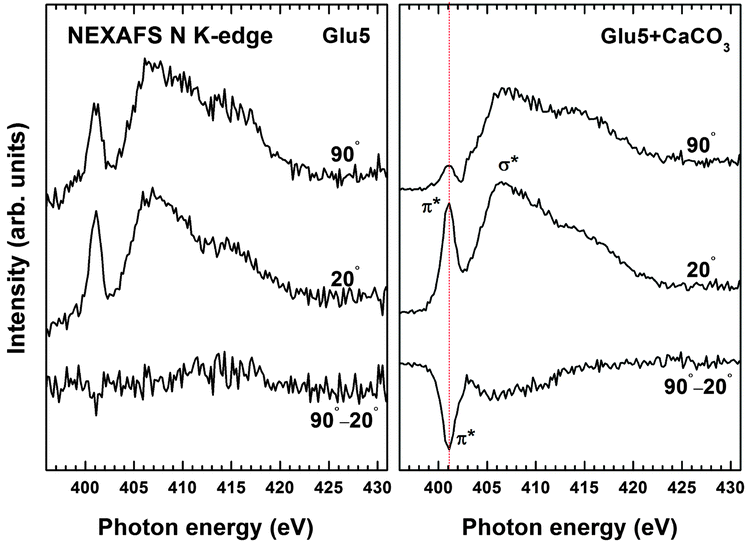 | ||
| Fig. 4 Normalized nitrogen K-edge NEXAFS spectra of Glu5 monolayers before (left) and after (right) growth of vaterite; spectra were acquired at X-ray incident angles of 90° and 20°, and are shown along with the difference spectra. The major resonances are marked in the spectra for Glu5 monolayer after growth of vaterite, and the dominant π* resonance is highlighted in the difference spectra. | ||
Apart from the electronic structure, the molecular orientation and order in the peptide can also be derived from the NEXAFS data, relying on the so-called linear dichroism, which can be described as the dependence of the absorption cross-section on the relative orientation of the molecular orbital and electric field vector of linearly polarized X-rays.29 The standard way of monitoring this linear dichroism is to analyse the difference between the spectra acquired at normal (90°) and grazing (20°) X-ray angles.29 The difference spectrum for the Glu5 SAM before exposure to the CaCO3 precursor is featureless, which indicates that the N–H and amide bonds in the peptide are largely disordered prior to mineralisation. After growth of vaterite on the peptide layer, however, an intense π* peak appears in the difference spectrum, which implies the backbone of the peptide became ordered during mineralisation. From further quantitative analysis, the average tilt angle of the π* molecular orbital can be calculated following the established theoretical framework.29 (see ESI† for fitting data) Assuming that Glu5 peptide forms a perfectly aligned monolayer and that the amide π* orbital is oriented perpendicular to the N–C–O plane of the peptide bond, then the Glu5 molecules tilt ∼35° with respect to the surface. Taking into account this tilt angle and the size of the peptide, we estimate a thickness of 15.2 Å for the Glu5 SAM, in good agreement with the XPS-derived thickness value of 15.9 Å (ESI†).
The induced structural order in the surface-bound Glu5 peptides upon mineralisation is also supported by SFG spectra recorded in the amide I region (Fig. 5). The SFG selection rules dictate that an SFG response will only originate from ordered species at a surface or interface,32,33i.e. in the present case, any vibrational mode observed will originate from ordered structures of the adsorbed peptide on Au.34 Despite the dense packing of the Glu5 molecules on Au as shown by the XPS data, no clear spectral features were observed in the SFG spectra of the pure Glu5 SAM, this indicates the surface-bound peptide largely lacks orientational order, behaving similarly as the random coils in bulk solution (see ESI† for circular dichroism spectra). In contrast, a clear SFG response is visible in the spectra after complexation with calcium ions, which is retained after growth of vaterite: two distinct bands at about 1535 cm−1 and 1680 cm−1, assigned to an asymmetric COO− side chain stretching mode35,36 and amide I mode within the peptide backbone,27,34,37 respectively. The amide I mode at ∼1680 cm−1 can be alternatively characteristic of 310 helices38,39 or ordered β-sheet structure.27,37 In the present case the latter structure is more probable since the 310 helix, having broad distribution of amide orientations within the helix, is not expected to contribute significantly to the angle dependence of amide peaks observed in the NEXAFS data. In addition, the more extended conformation of β-sheets matches the XPS and NEXAFS-derived film thickness result very well.
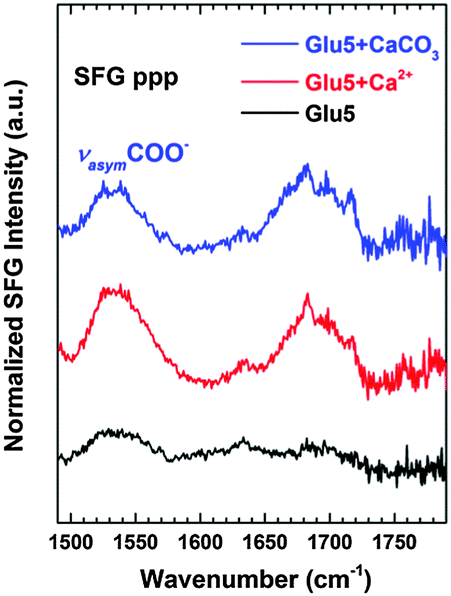 | ||
| Fig. 5 SFG amide I spectra of a Glu5 peptide SAM on Au (black curve), the peptide layer after complexation with calcium ions (red curve), and after growth of vaterite (blue). All the spectra were taken at ppp polarization. | ||
In the present study, the main driving force for vaterite formation is the strong interaction between surface-bound peptide and calcium ions: the abundant binding sites as well as high affinity between deprotonated peptide carboxyl groups and calcium ions will suppress the growth of calcite, thus favouring vaterite.13,16,17,19 Based on previous studies in solution,16,17 vaterite can only be stabilized by high peptide concentrations in the millimolar range. At the surface the local Glu5 peptide densities far exceed relevant solution state concentrations and can thereby very effectively stabilize vaterite. In addition to the inherent higher local concentration of carboxyl groups due to the surface immobilization,40 the restructuring of peptides obviously also plays a role: vaterite is formed by the cooperative interplay between Glu5 peptides and calcium ions (Fig. 6). The Glu5 molecules reduce their van der Waals ‘footprint’ on the surface upon interaction with ions, which results in ordered backbone and side chain structures; in this aligned state the peptides can serve as more effective structural templates for vaterite formation by providing a microenvironment with local supersaturation which, according to the Ostwald rule, leads to efficient nucleation of vaterite phase calcium carbonate. Here, calcium ions in the vaterite precursor solution act as the linking agent and structure the surface peptides, which, in turn, promote themselves into vaterite within the formed peptide template. In other words, calcium ions are ‘self-templating’ the formation of vaterite at the peptide–solution interface.
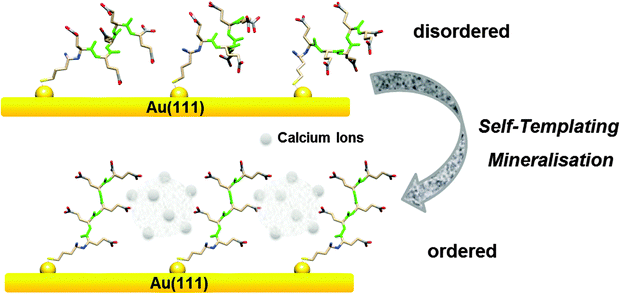 | ||
| Fig. 6 Schematic drawing of Glu5 peptide layer on Au when interacting with CaCO3 precursors. | ||
In summary, using the oligo(glutamic acid) peptides on gold as a model system, we demonstrated that vaterite CaCO3 can be stabilized by engineered peptides at surfaces. Calcium ions modify the peptide surface structure and thereby self-template vaterite structure at the surface. This finding provides important information about the interfacial biomineralisation of CaCO3, and indicates a biomineralisation pathway whereby template and precursor act together to trigger biomineralisation. Such a mechanism is possibly also relevant for other biogenic mineralisation phenomena.
We thank Gunnar Glasser, Jing Xie, Michael Steiert and Jennifer Heidrich for support of this work. HL and TW thank the European Commission (CIG grant #322124) and the Deutsche Forschungsgemeinschaft (WE4478/2-1) for financial support. We thank Daniel Fischer and Cherno Jaye (NIST) for providing us with the experimental equipment for NEXAFS spectroscopy and their help at the synchrotron. NEXAFS studies were performed at the NSLS, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Division of Materials Science and Division of Chemical Sciences.
Notes and references
- M. Suzuki, K. Saruwatari, T. Kogure, Y. Yamamoto, T. Nishimura, T. Kato and H. Nagasawa, Science, 2009, 325, 1388–1390 CrossRef CAS PubMed.
- J. Aizenberg, G. Lambert, S. Weiner and L. Addadi, J. Am. Chem. Soc., 2002, 124, 32–39 CrossRef CAS PubMed.
- Y. Levi, S. Albeck, A. Brack, S. Weiner and L. Addadi, Chem. – Eur. J., 1998, 4, 389–396 CrossRef CAS.
- A. M. Belcher, X. H. Wu, R. J. Christensen, P. K. Hansma, G. D. Stucky and D. E. Morse, Nature, 1996, 381, 56–58 CrossRef CAS PubMed.
- G. Falini, S. Albeck, S. Weiner and L. Addadi, Science, 1996, 271, 67–69 Search PubMed.
- J. Y. Xin, T. C. Chen, Z. F. Lin, P. Dong, H. Tan and J. S. Li, Chem. Commun., 2014, 50, 6491–6493 RSC.
- H. C. W. Skinner and H. Ehrlich, in Treatise on Geochemistry: Biogeochemistry, ed. H. D. Holland and K. K. Turekian, Elsevier Science, 2nd edn, 2014, vol. 10, pp. 105–162 Search PubMed.
- D. Wu, J. J. Yang, J. Y. Li, L. Chen, B. Tang, X. Y. Chen, W. Wu and J. S. Li, Biomaterials, 2013, 34, 5036–5047 CrossRef CAS PubMed.
- H. Ehrlich, Int. Geol. Rev., 2010, 52, 661–699 CrossRef PubMed.
- J. W. Morse, R. S. Arvidson and A. Luttge, Chem. Rev., 2007, 107, 342–381 CrossRef CAS PubMed.
- S. Kim and C. B. Park, Biomaterials, 2010, 31, 6628–6634 CrossRef CAS PubMed.
- K. Naka, Y. Tanaka and Y. Chujo, Langmuir, 2002, 18, 3655–3658 CrossRef CAS.
- D. B. Trushina, T. V. Bukreeva, M. V. Kovalchuk and M. N. Antipina, Mater. Sci. Eng., C, 2014, 45, 644–658 CrossRef CAS PubMed.
- A. L. Porter and W. J. Wilson, US Pat., CA2237960 A1, 1997 Search PubMed.
- M. Devenney, M. Fernandez and S. O. Morgan, US Pat., US 20130192783 A1, 2008 Search PubMed.
- M. A. Hood, K. Landfester and R. Muñoz-Espí, Cryst. Growth Des., 2014, 14, 1077–1085 CAS.
- B. Njegic-Dzakula, G. Falini, L. Brecevic, Z. Skoko and D. Kralj, J. Colloid Interface Sci., 2010, 343, 553–563 CrossRef CAS PubMed.
- P. Krattiger, N. Nassif, A. Volkel, Y. Mastai, H. Wennemers and H. Colfen, Colloids Surf., A, 2010, 354, 218–225 CrossRef CAS PubMed.
- H. Tong, W. Ma, L. Wang, P. Wan, J. Hu and L. Cao, Biomaterials, 2004, 25, 3923–3929 CrossRef CAS PubMed.
- H. Ehrlich, T. Hanke, P. Simon, R. Born, C. Fischer, A. Frolov, T. Langrock, R. Hoffmann, U. Schwarzenbolz, T. Henle, V. V. Bazhenov and H. Worch, J. Biomed. Mater. Res., Part B, 2010, 92b, 542–551 CAS.
- V. Fischer, K. Landfester and R. Muñoz-Espí, Cryst. Growth Des., 2011, 11, 1880–1890 CAS.
- H. A. Biebuyck, C. D. Bian and G. M. Whitesides, Langmuir, 1994, 10, 1825–1831 CrossRef CAS.
- M. Zharnikov, J. Electron Spectrosc. Relat. Phenom., 2010, 178, 380–393 CrossRef PubMed.
- P. E. Laibinis, G. M. Whitesides, D. L. Allara, Y. T. Tao, A. N. Parikh and R. G. Nuzzo, J. Am. Chem. Soc., 1991, 113, 7152–7167 CrossRef CAS.
- H. Hamoudi, K. Doring, F. Chesneau, H. Lang and M. Zharnikov, J. Phys. Chem. C, 2012, 116, 861–870 CAS.
- H. Lu, D. Zeysing, M. Kind, A. Terfort and M. Zharnikov, J. Phys. Chem. C, 2013, 117, 18967–18979 CAS.
- J. E. Baio, D. Schach, A. V. Fuchs, L. Schmuser, N. Billecke, C. Bubeck, K. Landfester, M. Bonn, M. Bruns, C. K. Weiss and T. Weidner, Chem. Commun., 2015, 51, 273–275 RSC.
- J. E. Baio, T. Weidner and D. G. Castner, Proteins at Interfaces III State of the Art, 2012, 1120, 761–779 CAS.
- J. Stöhr, NEXAFS Spectroscopy, Springer-Verlag, Berlin, 1992 Search PubMed.
- A. A. Makarova, E. V. Grachova, V. S. Neudachina, L. V. Yashina, A. Bluher, S. L. Molodtsov, M. Mertig, H. Ehrlich, V. K. Adamchuk, C. Laubschat and D. V. Vyalikh, Sci. Rep., 2015, 5, 8710 CrossRef CAS PubMed.
- T. Weidner, J. S. Apte, L. J. Gamble and D. G. Castner, Langmuir, 2010, 26, 3433–3440 CrossRef CAS PubMed.
- A. G. Lambert, P. B. Davies and D. J. Neivandt, Appl. Spectrosc. Rev., 2005, 40, 103–145 CrossRef CAS PubMed.
- Y. R. Shen, The Principles of Nonlinear Optics, J. Wiley, New York, 1984 Search PubMed.
- T. Weidner and D. G. Castner, Phys. Chem. Chem. Phys., 2013, 15, 12516–12524 RSC.
- I. A. Mudunkotuwa, A. A. Minshid and V. H. Grassian, Analyst, 2014, 139, 870–881 RSC.
- Z. Paszti and L. Guczi, Vib. Spectrosc., 2009, 50, 48–56 CrossRef CAS PubMed.
- J. E. Baio, A. Zane, V. Jaeger, A. M. Roehrich, H. Lutz, J. Pfaendtner, G. P. Drobny and T. Weidner, J. Am. Chem. Soc., 2014, 136, 15134–15137 CrossRef CAS PubMed.
- Y. W. Liu, J. Jasensky and Z. Chen, Langmuir, 2012, 28, 2113–2121 CrossRef CAS PubMed.
- S. J. Ye, K. T. Nguyen and Z. Chen, J. Phys. Chem. B, 2010, 114, 3334–3340 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details on sample preparation and characterization methods; XPS determined elemental composition for Glu5 SAM on Au; fitting result for N K-edge NEXAFS spectra of the Glu5 peptide after CaCO3 mineralisation. See DOI: 10.1039/c5cc05830a |
| ‡ Current address: Max Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. |
| This journal is © The Royal Society of Chemistry 2015 |