
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of diversely functionalised 2,2-disubstituted oxetanes: fragment motifs in new chemical space†
Owen A.
Davis
,
Rosemary A.
Croft
and
James A.
Bull
*
Department of Chemistry, Imperial College London, South Kensington, London SW7 2AZ, UK. E-mail: j.bull@imperial.ac.uk
First published on 27th August 2015
Abstract
Di-, tri- and tetra-substituted oxetane derivatives with combinations of ester, amide, nitrile, aryl, sulfone and phosphonate substituents are prepared as fragments or building blocks for drug discovery. The synthesis of these novel oxetane functional groups, in new chemical space, is achieved via rhodium-catalysed O–H insertion and C–C bond forming cyclisation.
Recent years has seen considerable enthusiasm for exploring new chemical motifs in medicinal chemistry that enter unexplored chemical and intellectual property (IP) space.1,2 Oxetanes motifs have gained significant traction as desirable modules for drug discovery.3 Carreira, Rogers-Evans, Müller and co-workers demonstrated that 3,3-disubstituted oxetanes constitute polar replacements for gem-dimethyl groups, and often afford improved solubility and physicochemical properties.4 Subsequently, oxetane motifs have been adopted widely in medicinal chemistry programs.5,6 3-Substituted oxetane motifs have been examined in particular, exploiting oxetan-3-one3,4 and 3-iodooxetane7 as building blocks. New spirocycles,8 peptidomimetics9 and other oxetane analogues of important compounds have been prepared (e.g.Fig. 1).6,10
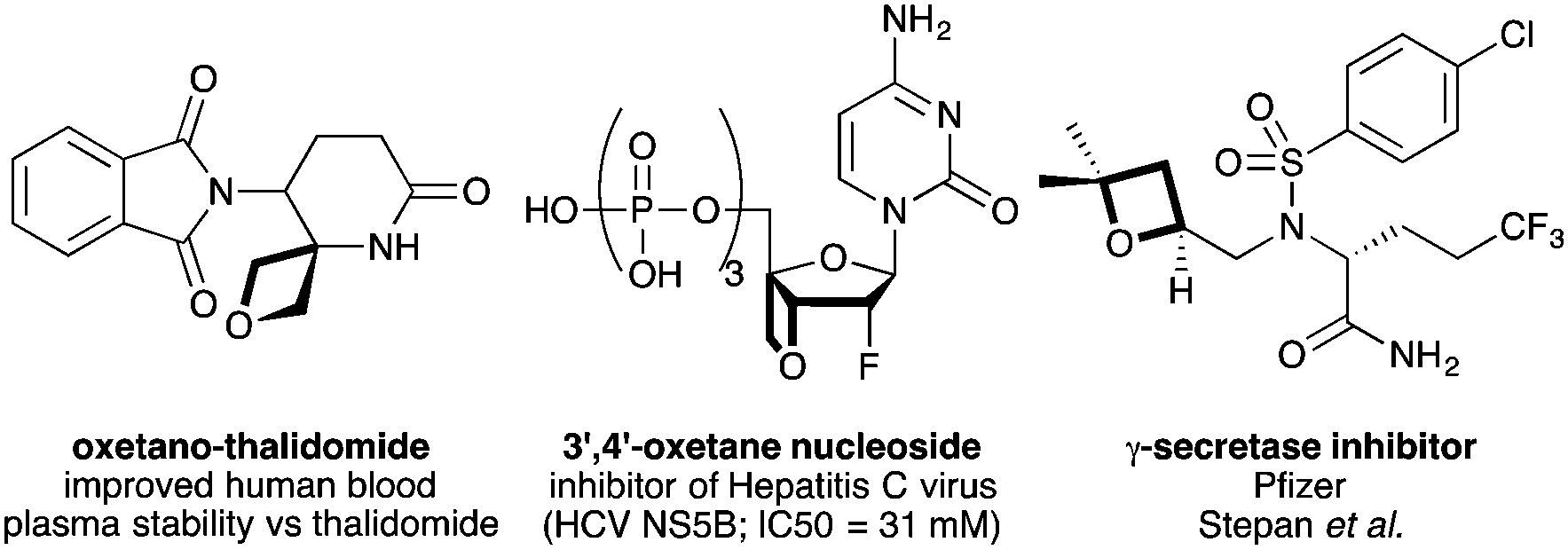 | ||
| Fig. 1 Biologically active oxetane-containing compounds. | ||
Examples of 2-substituted oxetanes remain less studied; Pfizer reported a class of 2-oxetanyl containing γ-secretase inhibitors that possessed improved properties relative to tetrahydrofuran and carbocyclic derivatives.6 However, oxetanes bearing functional groups on the ring are unexplored, and could provide fascinating new structural units for use by medicinal chemists. The synthesis of functionalised oxetanes continues to be a challenge. Cyclisation by intramolecular Williamson etherification is most common,3 but 2-functionality is often precluded due to unstable oxyanionic intermediates. Also, epoxide-opening and ring-closure methods using sulfoxonium ylides are not tolerant of sensitive electrophilic functionality.11 On the other hand, the photochemical Paterno–Büchi reaction can introduce functionality from an alkene component, but is often restricted to very highly substituted oxetanes, with undesirable properties for medicinal chemistry.12–14
We have targeted new oxetane derivatives due their small, polar nature and well-defined 3-D shape.15 We recently reported the preparation of 2-sulfonyl oxetanes, formed through a novel anionic C–C bond forming cyclisation (Scheme 1A).16 These fragments displayed good pH stability, and stability to liver microsomes.16b We have also established an efficient synthesis of oxetane 2,2-dicarboxylates involving O–H insertion using diazomalonates, and C–C bond forming cyclisation (Scheme 1B).17
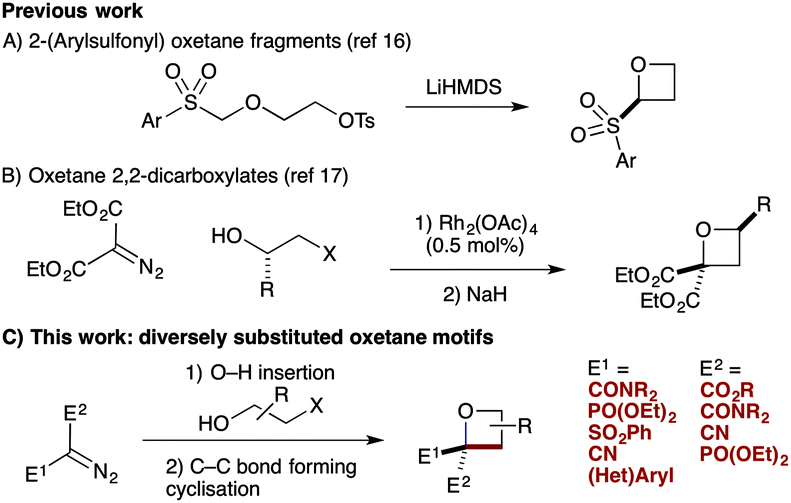 | ||
| Scheme 1 Synthesis of oxetanes through anionic C–C bond forming cyclisation. | ||
Here we report the synthesis of unusual oxetane motifs bearing medicinally relevant functional groups directly on the ring (Scheme 1C). Diversely functionalised oxetanes in new chemical space are prepared in a facile manner from functionalised diazo compounds by O–H insertion and C–C bond forming cyclisation. These simple but highly functionalised motifs provide interesting shape and physicochemical properties for fragments or building blocks for drug discovery that cannot be prepared by other methods.
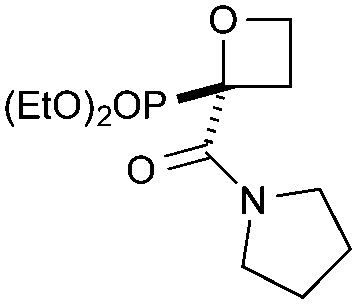
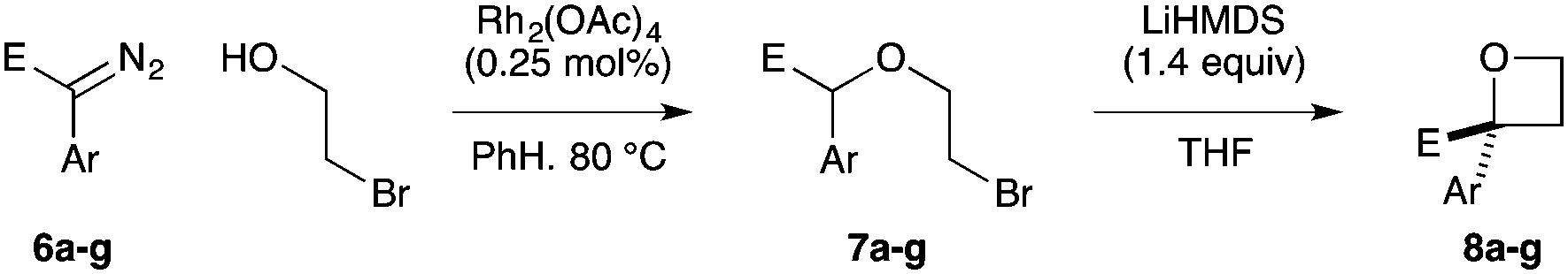
Targeting 2,2-difunctionalised oxetanes, our investigations initially focused on a range of unsymmetrical acceptor–acceptor diazo compounds, with which to examine O–H insertion with 2-bromoethanol (Scheme 1C and Table 1).18,19 A series of diazo compounds 1a–g bearing amide, sulfone, phosphonate, and nitrile groups were prepared from the methylene precursors using TsN3 or TfN3.18,20,21 We first examined amide–ester diazo 1a (entry 1). Pleasingly, using catalytic Rh2(OAc)4 (0.5 mol%) in benzene at 80 °C successfully mediated O–H insertion of the carbene derived from diazo 1a into 2-bromoethanol in 75% yield.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | E1 | E2 | Yield 2a (%) | Yield 3b (%) | Oxetane 3 | |
| a O–H insertion: 1 (1.1 equiv.), 2-bromoethanol (1.0–2.0 mmol), Rh2(OAc)4 (0.5 mol%), PhH, 0.1 M, 80 °C. b Cyclisation: 2 (0.3–0.6 mmol), NaH (1.2 equiv.), DMF, 0.025 M. c 0 °C for 1 h; then 25 °C for 1 h. d 0 °C for 1 h. e 1.5 equiv. diazo 1d. f Run in CH2Cl2, 0.1 M at 25 °C for 17 h. g 10 mmol scale. h 8 mmol scale. i 1.2 equiv. diazo 1f. | ||||||
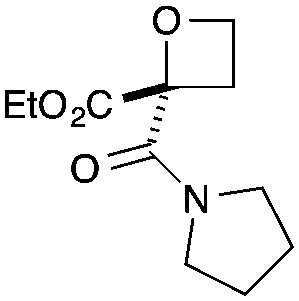
| 1 |
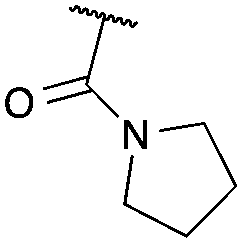
|
CO2Et | a | 75 | 93c |
|
| 2 |
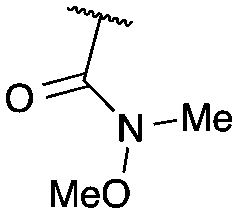
|
CO2Et | b | 51 | 96c |
|
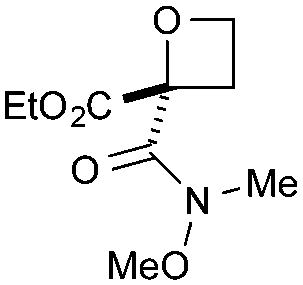
| 3 | SO2Ph | CO2Et | c | 92 | 87d |
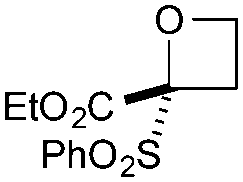
|
| 4 | PO(OEt)2 | CO2Et | d | 77e | 70d |
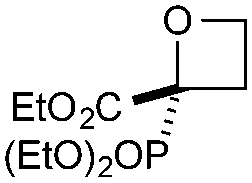
|
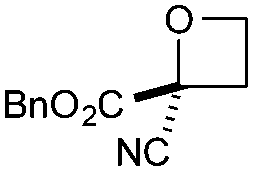
| 5 | CN | CO2Bn | e | 85f | 88d |
|
| 6 | 93f,g | 93d,h | ||||
| 7 |
|
CN | f | 67i | 87d |
|
| 8 |
|
PO(OEt)2 | g | 84 | 72d |
|
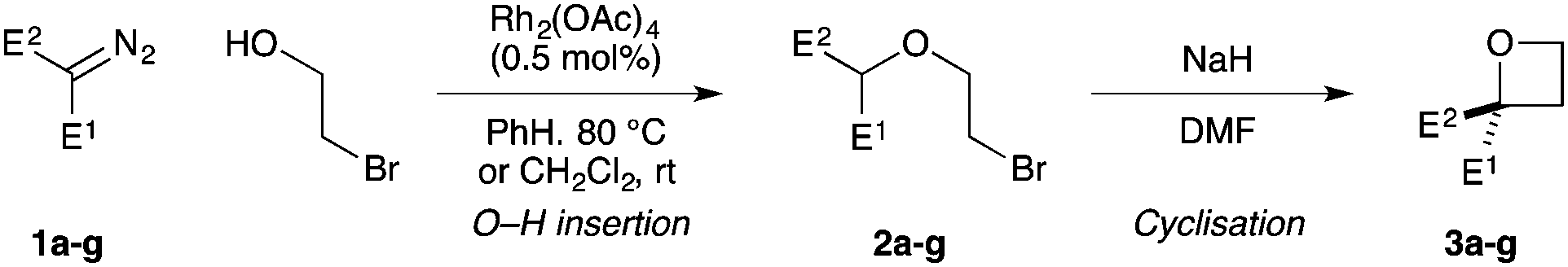
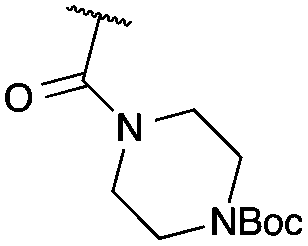
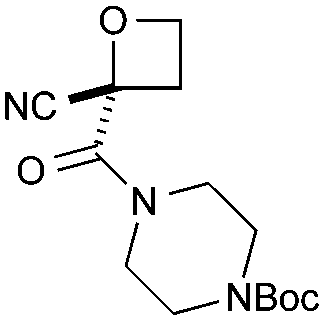
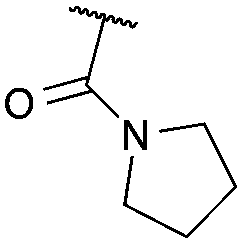
Cyclisation was then effected by treatment of 2a with NaH (1.2 equiv., 0 °C, 1 h, then 25 °C, 1 h) to afford 2-amido-2-ester-oxetane 3a in excellent yield forming the C(2)–C(3) bond. Using Weinreb amide 1b (entry 2), a slightly reduced yield was obtained for the O–H insertion (51%), but an excellent yield was obtained on cyclisation of 2b to oxetane 3b. Sulfone–ester diazo 1c smoothly underwent O–H insertion, followed by cyclisation in just 1 h at 0 °C to provide a new type of sulfonyl oxetane (entry 3).16 Phosphonate–ester diazo 1d proceeded similarly, to yield the first example of a 2-oxetanyl phosphonate, 3d.22 Interestingly, the use of nitrile–ester diazo 1e led to side reactions under the conditions for O–H insertion, with the carbenoid undergoing competitive insertion into the benzene solvent (1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–2.4:1 O–H insertion:π-insertion). To prevent this unwanted side product, the reaction was performed in CH2Cl2 (25 °C, 17 h), and bromide 2e was isolated in 85% yield (entry 5). Cyclisation at 0 °C afforded nitrile oxetane 3e after 1 h. The sequence was also successful on a larger scale affording >1.5 g of oxetane 3e in excellent yield (entry 6). Mixed nitrile–amide and phosphonate–amide diazo compounds were also successful under the same conditions, providing oxetanes 3f and 3g respectively (entries 7 and 8). The wide variety of substituted oxetanes that can be readily accessed demonstrates the versatility of this approach.
:1–2.4:1 O–H insertion:π-insertion). To prevent this unwanted side product, the reaction was performed in CH2Cl2 (25 °C, 17 h), and bromide 2e was isolated in 85% yield (entry 5). Cyclisation at 0 °C afforded nitrile oxetane 3e after 1 h. The sequence was also successful on a larger scale affording >1.5 g of oxetane 3e in excellent yield (entry 6). Mixed nitrile–amide and phosphonate–amide diazo compounds were also successful under the same conditions, providing oxetanes 3f and 3g respectively (entries 7 and 8). The wide variety of substituted oxetanes that can be readily accessed demonstrates the versatility of this approach.
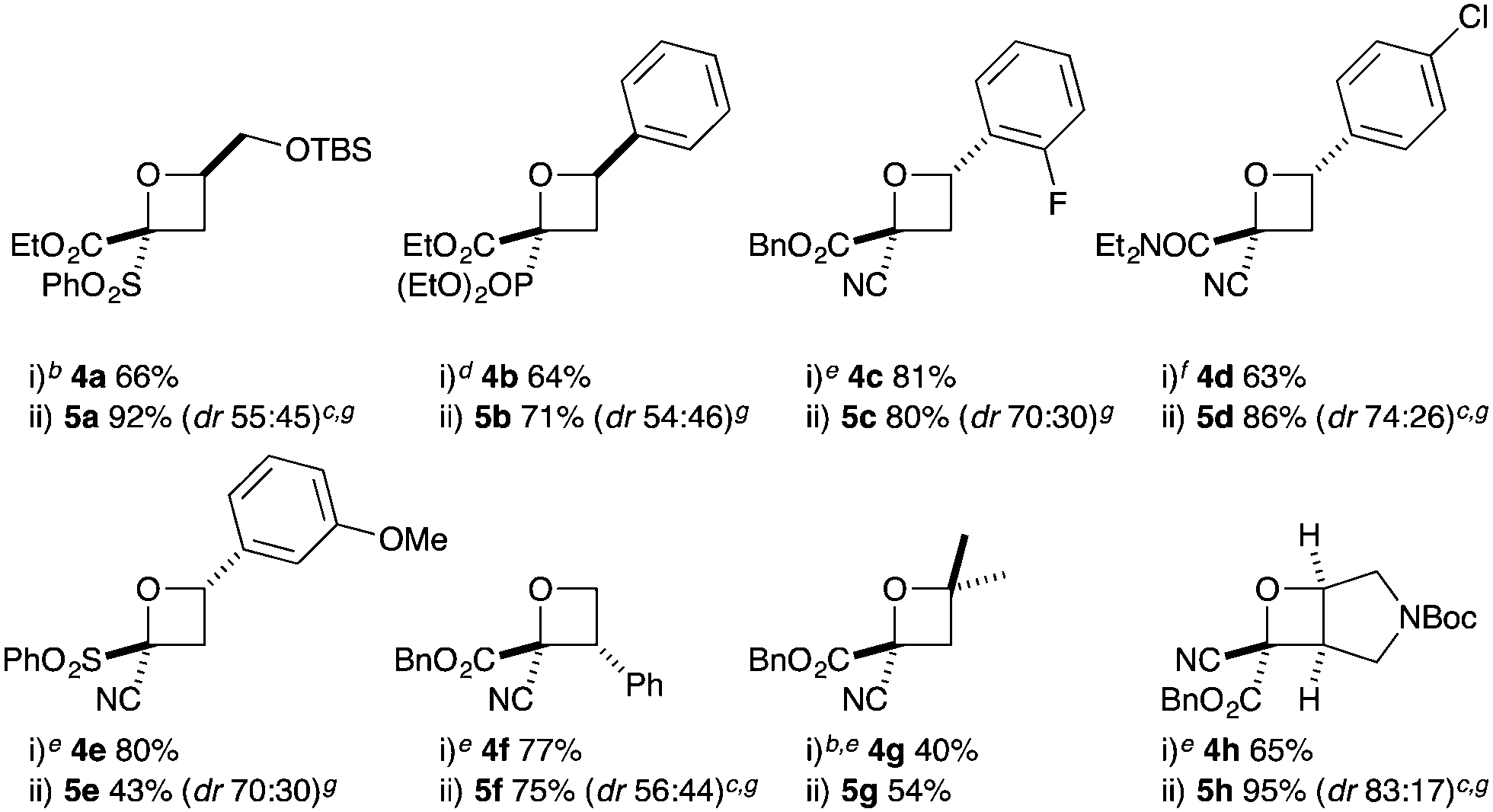
Substituted bromohydrins were then investigated to form tri- and tetrasubstituted oxetane derivatives (Table 2). The reaction sequence successfully formed oxetanes with 4-alkyl (5a) and 4-aryl substituents (5b–e) in good yields using similar reaction conditions. The additional substituent had little influence on the O–H insertion step. However, elevated temperature and increased reaction times were required for cyclisation of all substituted examples, requiring 16 h at 25 °C for complete conversion.
|
|
|---|
| Conditions: (i) O–H insertion: 1 (1.1 equiv.), β-bromohydrin (0.5–1.3 mmol), Rh2(OAc)4 (0.5 mol%), PhH, 0.1 M, 80 °C. (ii) Cyclisation: 4 (0.3–0.5 mmol), NaH (1.2 equiv.), DMF, 0.025 M, 25 °C, 16 h.a dr determined by 1H NMR of crude reaction mixture.b β-Chlorohydrin used.c Oxetane isomers separated by silica chromatography.d 1.5 equiv. diazo.e Run in CH2Cl2, 0.1 M at 25 °C for 17 h.f 1.2 equiv. diazo.g Major diastereoisomer indicated (see ref. 23). |
|
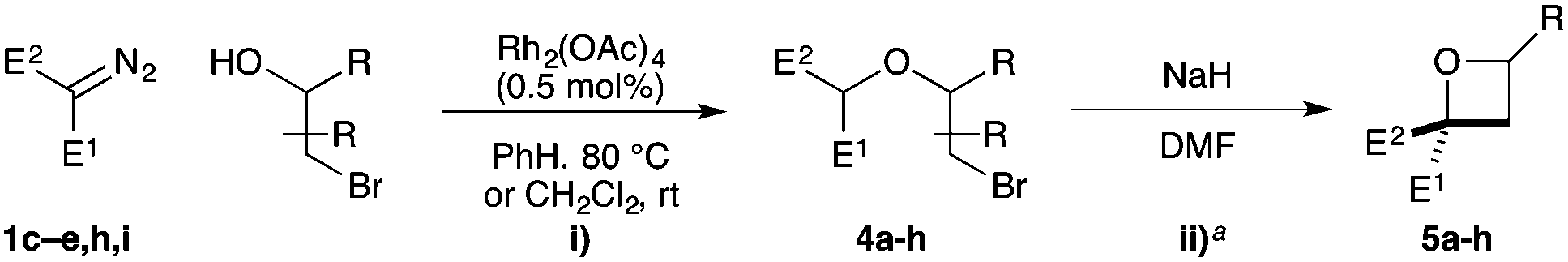
For oxetanes 5a and 5b, a ∼1:1 mixture of diastereoisomers was obtained due to the sterically similar ester–sulfone or ester–phosphonate groups.23 Diazo compounds with a less sterically demanding nitrile group (1e, 1h, 1i) gave increased diastereoselectivity (up to 3:1 dr). The incorporation of a 3-Ph group gave the 2,2,3-substituted derivative 5f (dr = 56:44). 2,2,4,4-Tetrasubstituted oxetane 5g was prepared from the commercially available chlorohydrin (1-chloro-2-methyl-2-propanol) in moderate yields. The synthesis of novel fused bicyclic oxetane scaffold 5h started by treating N-Boc-3-pyrroline with NBS and H2O to provide 3-bromo-4-hydroxy-pyrrolidine, which gave ether 4h on O–H insertion. Cyclisation then afforded the bicyclic oxetane 5h as a 83:17 mixture of separable diastereoisomers in 95% combined yield.21,23
Next we examined aryl–ester (donor–acceptor) diazo compounds,24 to generate 2-aryl-oxetane-2-carboxylates (Table 3). Such aryl-substituted heterocycles offer attractive features for fragment screening and are little studied in medicinal chemistry.25 To our delight, the O–H insertion conditions were effective for the phenyl–ester diazo 6a with an excellent 86% yield using reduced Rh2(OAc)4 loading (0.25 mol%, entry 1). The O–H insertion was successful across varied aryl groups (entries 3–8), including the chloropyridyl diazo 6f. However, using the NaH in DMF conditions for cyclisation gave variable yields and oxetane 8a and bromide 7a proved inseparable. Alternative bases and conditions were examined,21 and the use of LiHMDS (1.4 equiv.) in THF was found to give quantitative conversion to oxetane 8a with a 93% isolated yield (entry 1). This reaction sequence was similarly successful on a larger scale (entry 2). The modified cyclisation conditions worked well for substituted aryl derivatives including halogenated (entries 4 and 5), and pyridyl derivatives (entry 8). However, the electron rich p-methoxy phenyl substrate (entry 6) led to low conversion and isolated yield. Using a larger excess of LiHMDS (2 equiv.) gave an improved yield of 41% (entry 7). The cyclisation conditions were also effective for aryl–amide ether 7g (entry 9). This approach provided direct access to compounds that conform well to fragment guidelines for fragment screening.21
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ar | E | Yield 7a (%) | Yield 8b (%) | Oxetane 8 | |
| a O–H insertion: 6 (1.1–1.5 equiv.), 2-bromoethanol (0.4–2.0 mmol), Rh2(OAc)4 (0.25 mol%), PhH, 0.1 M, 80 °C. b Cyclisation: 7 (0.3–0.5 mmol), LiHMDS (1.4 equiv.), THF, 0.025 M, 0 °C, 1 h. c Reaction on 7.5 mmol scale. d Reaction on 5.0 mmol scale. e 0.5 mol% Rh2(OAc)4. f 2.0 equiv. LiHMDS. | ||||||
| 1 | Ph | CO2Et | a | 86 | 93 |
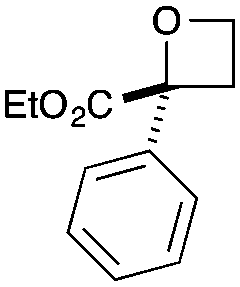
|
| 2 | 80c | 76d | ||||
| 3 | Ph | CO2iPr | b | 87e | 72 |
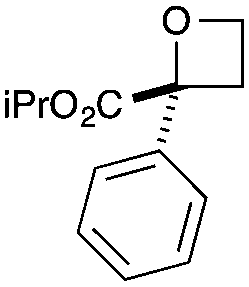
|
| 4 | 3-ClPh | CO2Et | c | 69 | 97 |
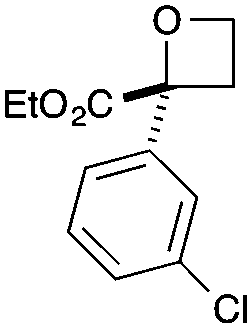
|
| 5 | 4-CF3Ph | CO2Et | d | 64 | 77 |
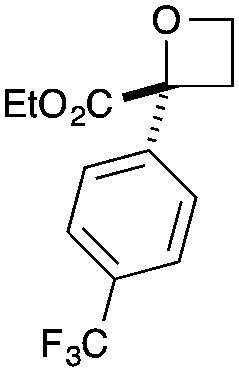
|
| 6 | 4-MeOPh | CO2Et | e | 86 | 22 |
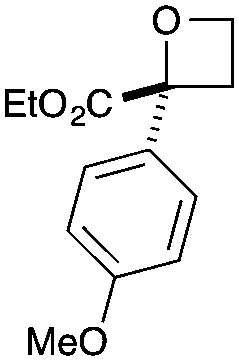
|
| 7 | 41f | |||||
| 8 |
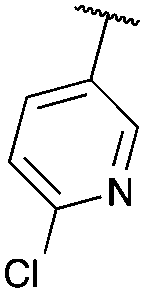
|
CO2Et | f | 54 | 76 |
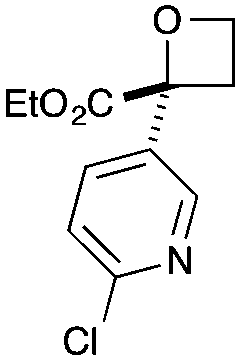
|
| 9 | 3-ClPh |
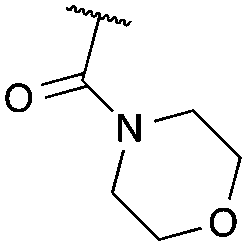
|
g | 73 | 90 |
|
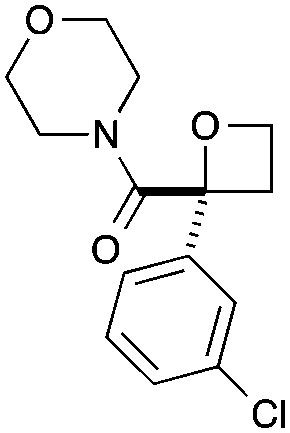
As a more divergent approach to similar compounds, hydrolysis of ester oxetane 8a was examined (Scheme 2a). Using NaOH afforded sodium salt 9 as an attractive oxetane containing building block. Amide coupling with morpholine, directly from the salt,26 gave amide 10 in a good yield over two steps. Finally, we prepared additional 2-nitrile oxetane derivatives. The nitrile group is receiving considerable attention in drug discovery,27,28 being metabolically robust and a strong hydrogen bond acceptor. Therefore, we considered that the combination with an oxetane motif could present a fascinating structure for medicinal chemistry.13,29 From nitrile–oxetane benzyl ester 3e, ester hydrolysis was best achieved using LiOH to afford lithium salt 11 (Scheme 2b). Amide bond formation gave nitrile amide oxetanes 12 and 13 in 30–35% yields over 2 steps. Importantly, treatment of oxetane 3f with TFA promoted Boc deprotection, 14, without other degradation, providing encouraging preliminary information on acid stability (Scheme 2c; 10 equiv. TFA, 22 h).
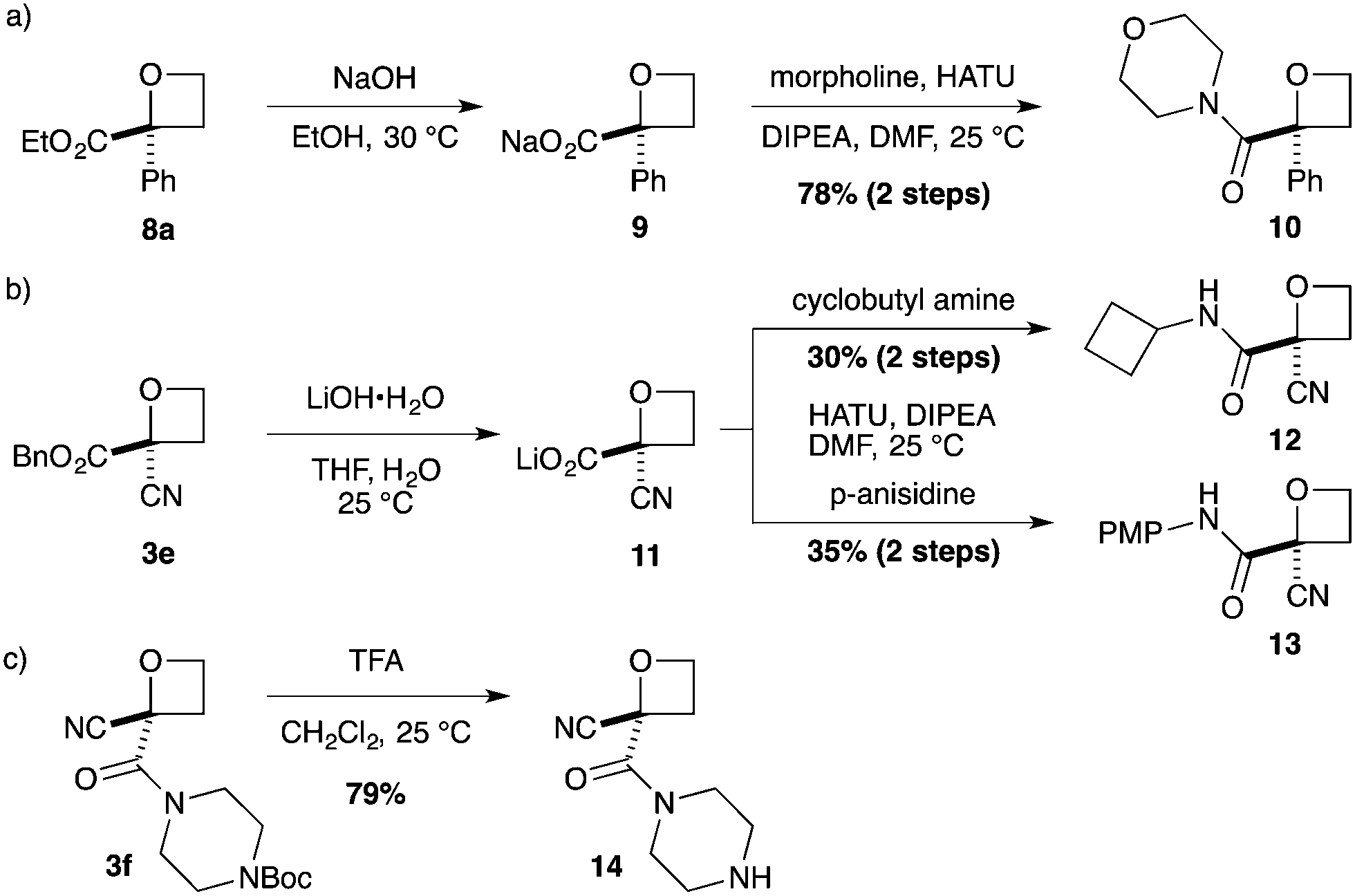 | ||
| Scheme 2 Divergent synthesis of oxetane derivatives. | ||
In summary, we have developed a versatile strategy to prepare diversely functionalised oxetane derivatives, involving O–H insertion and C–C bond forming cyclisation. A wide variety of functional groups have been introduced on the oxetane ring, accessing new chemical space. We believe these oxetane motifs will provide interesting new structural elements for medicinal chemistry programs as well as synthesis. Efforts towards enantioselective synthesis and studies on the stability and medicinal chemistry properties of the new oxetane derivatives will be reported in due course. We gratefully acknowledge EPSRC (CAF, EP/J001538/1; Impact Acceleration Account, EP/K503733/1), and Imperial College London. We thank EPSRC National Mass Spectrometry Facility, Swansea.
Notes and references
- (a) J.-L. Reymond, R. van Deursen, L. C. Blum and L. Ruddigkeit, Med. Chem. Commun., 2010, 1, 30 RSC; (b) R. J. Hall, P. N. Mortenson and C. W. Murray, Prog. Biophys. Mol. Biol., 2014, 116, 82 CrossRef CAS; (c) D. H. Drewry and R. Macarron, Curr. Opin. Chem. Biol., 2010, 14, 289 CrossRef CAS; (d) C. M. Marson, Chem. Soc. Rev., 2011, 40, 5514 RSC; (e) W. R. Pitt, D. M. Parry, B. G. Perry and C. R. Groom, J. Med. Chem., 2009, 52, 2952 CrossRef CAS; (f) F. W. Goldberg, J. G. Kettle, T. Kogej, M. W. D. Perry and N. P. Tomkinson, Drug Discovery Today, 2015, 20, 11 CrossRef.
- For examples: (a) J. A. Burkhard and E. M. Carreira, Org. Lett., 2008, 10, 3525 CrossRef CAS; (b) J. A. Burkhard, C. Guérot, H. Knust, M. Rogers-Evans and E. M. Carreira, Org. Lett., 2010, 12, 1944 CrossRef CAS; (c) J. A. Burkhard, B. Wagner, H. Fischer, F. Schuler, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 3524 CrossRef CAS; (d) D. B. Li, M. Rogers-Evans and E. M. Carreira, Org. Lett., 2011, 13, 6134 CrossRef CAS; (e) D. Foley, R. Doveston, I. Churcher, A. Nelson and S. P. Marsden, Chem. Commun., 2015, 51, 11174 RSC.
- J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052 CrossRef CAS.
- (a) G. Wuitschik, M. Rogers-Evans, K. Müller, H. Fischer, B. Wagner, F. Schuler, L. Polonchuk and E. M. Carreira, Angew. Chem., Int. Ed., 2006, 45, 7736 CrossRef CAS; (b) G. Wuitschik, E. M. Carreira, B. Wagner, H. Fischer, I. Parrilla, F. Schuler, M. Rogers-Evans and K. Müller, J. Med. Chem., 2010, 53, 3227 CrossRef CAS.
- (a) J. Du, B.-K. Chun, R. T. Mosley, S. Bansal, H. Bao, C. Espiritu, A. M. Lam, E. Murakami, C. Niu, H. M. Micolochick Steuer, P. A. Furman and M. J. Sofia, J. Med. Chem., 2014, 57, 1826 CrossRef CAS; (b) E. M. Skoda, J. R. Sacher, M. Z. Kazancioglu, J. Saha and P. Wipf, ACS Med. Chem. Lett., 2014, 5, 900 CrossRef CAS; (c) J. E. Dowling, M. Alimzhanov, L. Bao, M. H. Block, C. Chuaqui, E. L. Cooke, C. R. Denz, A. Hird, S. Huang, N. A. Larsen, B. Peng, T. W. Pontz, C. Rivard-Costa, J. C. Saeh, K. Thakur, Q. Ye, T. Zhang and P. D. Lyne, ACS Med. Chem. Lett., 2013, 4, 800 CrossRef CAS.
- (a) A. F. Stepan, K. Karki, W. S. McDonald, P. H. Dorff, J. K. Dutra, K. J. DiRico, A. Won, C. Subramanyam, I. V Efremov, C. J. O'Donnell, C. E. Nolan, S. L. Becker, L. R. Pustilnik, B. Sneed, H. Sun, Y. Lu, A. E. Robshaw, D. Riddell, T. J. O'Sullivan, E. Sibley, S. Capetta, K. Atchison, A. J. Hallgren, E. Miller, A. Wood and R. S. Obach, J. Med. Chem., 2011, 54, 7772 CrossRef CAS; (b) A. F. Stepan, G. W. Kauffman, C. E. Keefer, P. R. Verhoest and M. Edwards, J. Med. Chem., 2013, 56, 6985 CrossRef CAS.
- (a) M. A. J. Duncton, M. A. Estiarte, R. J. Johnson, M. Cox, D. J. R. O'Mahony, W. T. Edwards and M. G. Kelly, J. Org. Chem., 2009, 74, 6354 CrossRef CAS; (b) M. A. J. Duncton, M. A. Estiarte, D. Tan, C. Kaub, D. J. R. O'Mahony, R. J. Johnson, M. Cox, W. T. Edwards, M. Wan, J. Kincaid and M. G. Kelly, Org. Lett., 2008, 10, 3259 CrossRef CAS.
- (a) J. A. Burkhard, C. Guérot, H. Knust and E. M. Carreira, Org. Lett., 2012, 14, 66 CrossRef CAS; (b) G. Wuitschik, M. Rogers-Evans, A. Buckl, M. Bernasconi, M. Märki, T. Godel, H. Fischer, B. Wagner, I. Parrilla, F. Schuler, J. Schneider, A. Alker, W. B. Schweizer, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4512 CrossRef CAS.
- (a) N. H. Powell, G. J. Clarkson, R. Notman, P. Raubo, N. G. Martin and M. Shipman, Chem. Commun., 2014, 50, 8797 RSC; (b) M. McLaughlin, R. Yazaki, T. C. Fessard and E. M. Carreira, Org. Lett., 2014, 16, 4070 CrossRef CAS.
- For oxetano-thalidomide, see: (a) J. A. Burkhard, G. Wuitschik, J.-M. Plancher, M. Rogers-Evans and E. M. Carreira, Org. Lett., 2013, 15, 4312 CrossRef CAS; (b) for nucleoside analogue: W. Chang, J. Du, S. Rachakonda, B. S. Ross, S. Convers-Reignier, W. T. Yau, J.-F. Pons, E. Murakami, H. Bao, H. M. Steuer, P. A. Furman, M. J. Otto and M. J. Sofia, Bioorg. Med. Chem. Lett., 2010, 20, 4539 CrossRef CAS.
- (a) T. Sone, G. Lu, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2009, 48, 1677 CrossRef CAS; (b) E. D. Butova, A. V. Barabash, A. A. Petrova, C. M. Kleiner, P. R. Schreiner and A. A. Fokin, J. Org. Chem., 2010, 75, 6229 CrossRef CAS.
- (a) M. Abe, J. Chin. Chem. Soc., 2008, 55, 479 CrossRef CAS; (b) F. Vogt, K. Jödicke, J. Schröder and T. Bach, Synthesis, 2009, 4268 CAS.
- For Paterno–Büchi reactions with 1,2-dicyanoethylene, forming nitrile oxetanes, see: (a) W. S. Chung, N. J. Turro, S. Srivastava and W. J. Le Noble, J. Org. Chem., 1991, 56, 5020 CrossRef CAS; (b) C. W. Funke and H. Cerfontain, J. Chem. Soc., Perkin Trans. 1, 1976, 1902 RSC; (c) J. A. Barltrop and H. A. J. Carless, J. Am. Chem. Soc., 1972, 94, 1951 CrossRef CAS.
- For 2,2-disubstituted oxetanes by lithiation of phenyl oxetane, see: D. I. Coppi, A. Salomone, F. M. Perna and V. Capriati, Chem. Commun., 2011, 47, 9918 RSC.
- For interest in less-planar compounds in drug discovery: (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS; (b) M. Ishikawa and Y. Hashimoto, J. Med. Chem., 2011, 54, 1539 CrossRef CAS; (c) F. Lovering, Med. Chem. Commun., 2013, 4, 515 RSC; (d) A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114 CrossRef CAS.
- (a) K. F. Morgan, I. A. Hollingsworth and J. A. Bull, Chem. Commun., 2014, 50, 5203 RSC; (b) K. F. Morgan, I. A. Hollingsworth and J. A. Bull, Org. Biomol. Chem., 2015, 13, 5265 RSC.
- (a) O. A. Davis and J. A. Bull, Angew. Chem., Int. Ed., 2014, 53, 14230 CrossRef CAS; (b) O. A. Davis and J. A. Bull, Synlett, 2015, 1283 CAS.
- For extensive studies on O–H insertion with varied diazo compounds, see: (a) G. G. Cox, D. J. Miller, C. J. Moody, E.-R. H. B. Sie and J. J. Kulagowski, Tetrahedron, 1994, 50, 3195 CrossRef CAS; (b) C. J. Moody, E. R. H. B. Sie and J. J. Kulagowski, Tetrahedron, 1992, 48, 3991 CrossRef CAS; (c) for a review: D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918 RSC.
- For an O–H insertion on bromoethanol, see: U. Trstenjak, J. Ilaš and D. Kikelj, Tetrahedron Lett., 2013, 54, 3341 CrossRef CAS.
- (a) J. C. Lee and J. Y. Yuk, Synth. Commun., 1995, 25, 1511 CrossRef CAS; (b) D. Marcoux, S. Azzi and A. B. Charette, J. Am. Chem. Soc., 2009, 131, 6970 CrossRef CAS; (c) R. P. Wurz, W. Lin and A. B. Charette, Tetrahedron Lett., 2003, 44, 8845 CrossRef CAS; (d) D. Marcoux and A. B. Charette, Angew. Chem., Int. Ed., 2008, 47, 10155 CrossRef CAS.
- See ESI† for further details.
- For an example of a highly substituted 2-phosponate-oxetan-3-one: T. Maegawa, K. Otake, K. Hirosawa, A. Goto and H. Fujioka, Org. Lett., 2012, 14, 4798 CrossRef CAS.
- Major diastereoisomers shown determined by nOe studies (5a, 5d, 5f, 5h) or by analogy. See ESI† for further details.
- Aryl ester diazo compounds prepared using pABSA reagent. (a) H. M. L. Davies, W. R. Cantrell Jr., K. R. Romines and J. S. Baum, Org. Synth., 1992, 70, 93 CrossRef CAS; (b) H. M. L. Davies, M. V. A. Grazini and E. Aouad, Org. Lett., 2001, 3, 1475 CrossRef CAS.
- For an isolated report of related structure, see: E. K. Bayburt, B. Clapham, P. B. Cox, J. F. Daanen, M. J. Dart, G. A. Gfesser, A. Gomtsyan, M. E. Kort, P. R. Kym, R. G. Schmidt and E. A. Voight, Novel trpv3 modulators, US Pat., US2013/0131036 A1, 2013 Search PubMed.
- J. D. Goodreid, P. A. Duspara, C. Bosch and R. A. Batey, J. Org. Chem., 2014, 79, 943 CrossRef CAS.
- For a review of nitrile containing pharmaceuticals, see: F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef CAS.
- For anastrazole: (a) M. Milani, G. Jha and D. A. Potter, Clin. Med.: Ther., 2009, 1, 141 CAS; (b) T. E. Hughes, M. D. Mone, M. E. Russell, S. C. Weldon and E. B. Villhauer, Biochemistry, 1999, 38, 11597 CrossRef CAS.
- For other nitrile oxetanes: (a) P. Sulmon, N. De Kimpe, N. Schamp, B. Tinant and J.-P. Declercq, Tetrahedron, 1988, 44, 3653 CrossRef CAS; (b) T. S. Cantrell, J. Chem. Soc., Chem. Commun., 1975, 637 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data, 1H and 13C NMR spectra; details of reaction optimisation and calculated molecular properties. See DOI: 10.1039/c5cc05740j |
| This journal is © The Royal Society of Chemistry 2015 |