
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceortho-(Methyltosylaminoethynyl)benzyl glycosides as new glycosyl donors for latent-active glycosylation†
Xiaoping
Chen
a,
Dacheng
Shen
a,
Qiaoling
Wang
a,
You
Yang
b and
Biao
Yu
*a
aState Key Laboratory of Bio-organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: byu@mail.sioc.ac.cn
bShanghai Key Laboratory of New Drug Design, School of Pharmacy, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
First published on 27th July 2015
Abstract
A new glycosylation protocol employing ortho-(methyltosylaminoethynyl)benzyl glycosides as glycosyl donors and TMSOTf as the catalyst is disclosed. These donors can be readily prepared from the corresponding ‘latent’ ortho-iodobenzyl glycosides via a Sonogashira coupling, thus providing a new approach for the ‘latent-active’ synthesis of glycans.
New glycosylation methods are continuously developed with efforts to construct the extremely diverse glycosidic linkages occurring in glycans and glycoconjugates in an efficient and economical manner.1 In the last decade, much interest has been devoted to the investigation of glycosylation protocols based on activation of the C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bonds. Thus, propargyl glycosides and 1,2-orthoesters (under the catalysis of AuX3),2,3 glycosyl alkynoates (under the promotion of Hg(OTf)2),4 dipropargylcyanoacetates (under the catalysis of AuCl3/AgSbF6),5 as well as alkynyl-containing thioglycosides (under the catalysis of Au(I) complexes),6 are disclosed to be effective donors in certain glycosylation reactions. Especially, glycosyl ortho-alkynylbenzoates (under the catalysis of a gold(I) complex, such as PPh3AuNTf2 and PPh3AuOTf) have been found to have wide applications in the synthesis of complex glycans and glycoconjugates;7,8 and the general mechanism of this glycosylation reaction has been largely elucidated.7c–e During the course of these studies, we tried glycosyl ortho-alkynylbenzyl glycosides as donors, which would have the advantage of easy manipulation of the protecting groups, but found no glycosylation took place under similar conditions wherein the corresponding ortho-alkynylbenzoates underwent glycosylation. We envisioned introduction of an electron-rich substituent on the alkyne moiety to facilitate the desired glycosylation pathway. Here we report ortho-(methyltosylaminoethynyl)benzyl glycosides as a new type of glycosyl donors which can be activated by a catalytic amount of TMSOTf under mild conditions and their applicability in the ‘latent-active’ synthesis of glycans.
C triple bonds. Thus, propargyl glycosides and 1,2-orthoesters (under the catalysis of AuX3),2,3 glycosyl alkynoates (under the promotion of Hg(OTf)2),4 dipropargylcyanoacetates (under the catalysis of AuCl3/AgSbF6),5 as well as alkynyl-containing thioglycosides (under the catalysis of Au(I) complexes),6 are disclosed to be effective donors in certain glycosylation reactions. Especially, glycosyl ortho-alkynylbenzoates (under the catalysis of a gold(I) complex, such as PPh3AuNTf2 and PPh3AuOTf) have been found to have wide applications in the synthesis of complex glycans and glycoconjugates;7,8 and the general mechanism of this glycosylation reaction has been largely elucidated.7c–e During the course of these studies, we tried glycosyl ortho-alkynylbenzyl glycosides as donors, which would have the advantage of easy manipulation of the protecting groups, but found no glycosylation took place under similar conditions wherein the corresponding ortho-alkynylbenzoates underwent glycosylation. We envisioned introduction of an electron-rich substituent on the alkyne moiety to facilitate the desired glycosylation pathway. Here we report ortho-(methyltosylaminoethynyl)benzyl glycosides as a new type of glycosyl donors which can be activated by a catalytic amount of TMSOTf under mild conditions and their applicability in the ‘latent-active’ synthesis of glycans.
The desired glycosyl ortho-(methyltosylaminoethynyl)benzyl glycosides could be easily prepared from the corresponding ortho-iodobenzyl glycosides via a Sonogashira coupling with ynamide 4.9–11 Taking the preparation of perbenzoyl glucopyranoside 1a as an example (Scheme 1), ortho-iodobenzyl glucopyranoside 3a was obtained via condensation of perbenzoylated glucose 2a with 2-iodobenzyl alcohol under the action of TMSOTf, which was then subjected to coupling with ynamide 4 in the presence of (PPh3)2PdCl2 and CuI in i-Pr2NH/DMF to provide the desired ortho-(methyltosylaminoethynyl)benzyl glucoside 1a in high yield (90%).10 Manipulation of the protecting groups on the iodobenzyl glycoside 3a followed by Sonogashira coupling would lead to (methyltosylaminoethynyl)benzyl glucopyranosides bearing different protecting group patterns, such as 1c (Table 2). (Methyltosylaminoethynyl)benzyl rhamnopyranoside 1b and 2-deoxy-glucopyranoside 1d were similarly prepared by Sonogashira coupling as the key step (see ESI† for details). All these glycosides were found to be stable when stored at room temperature.
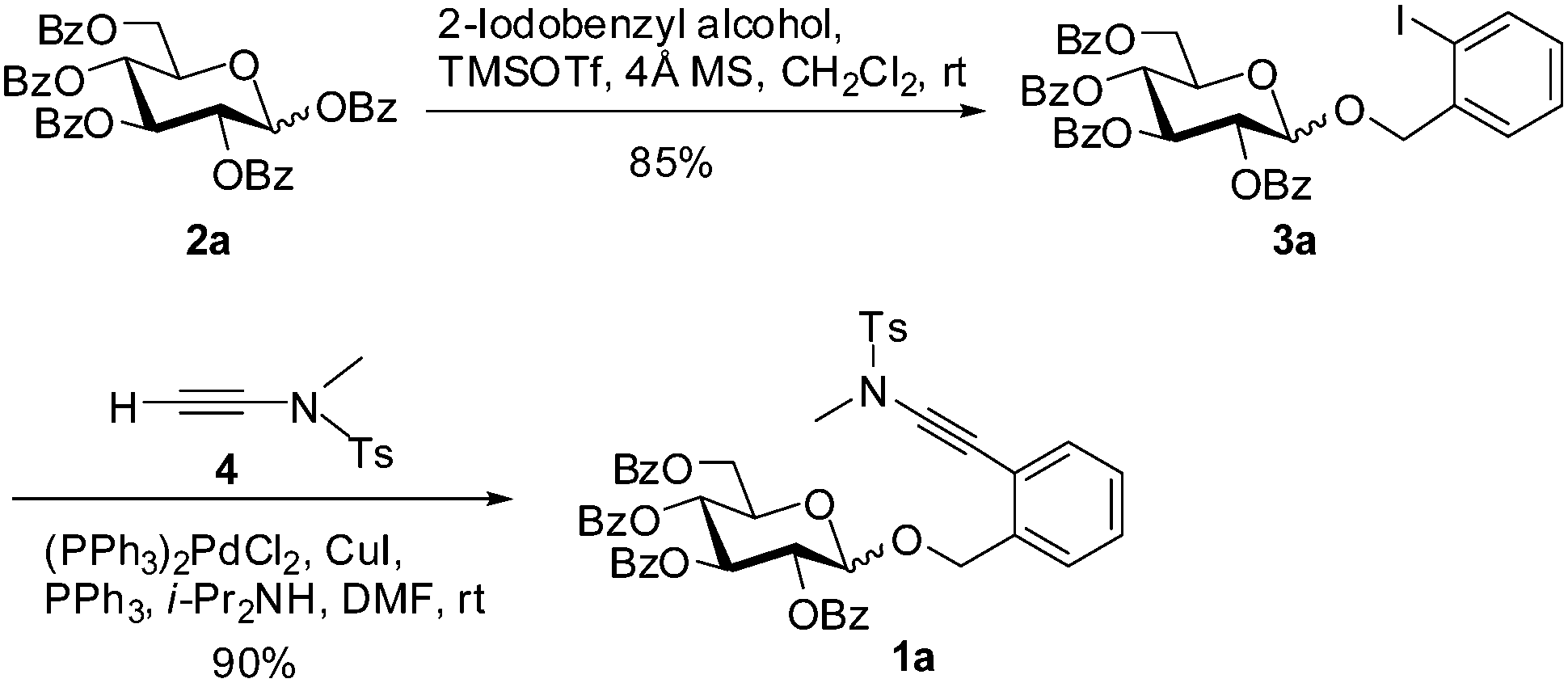 | ||
| Scheme 1 Preparation of ortho-(methyltosylaminoethynyl)benzyl glucopyranoside 1a. | ||
With the perbenzoyl glucoside 1a as a potential donor and cholesterol 5a as an acceptor, a variety of π-acids and Lewis acids (0.1 eq.) were screened as promoters for the desired glycosylation reaction in the presence of 4 Å MS in CH2Cl2 at room temperature (Table 1). Surprisingly, PPh3AuNTf2, the effective catalyst for the glycosylation of glycosyl ortho-alkynylbenzoates,7 could not catalyze the present coupling effectively, providing the desired β-glucoside 6aa in only 43% yield (entry 1). The major by-product arose from the nucleophilic addition of cholesterol 5a onto the ynamide moiety.11a,e TMSOTf turned out to be the most effective catalyst, wherein the coupled glycoside 6aa was obtained in a high yield of 91% (entry 2). Bi(OTf)3, In(OTf)3, and Cu(OTf)2 were shown to be better catalysts than PPh3AuNTf2, leading to 6aa in 83%, 67%, and 46% yield, respectively (entries 3–5). Sc(OTf)3, BF3OEt2, SnCl4, and PtCl2 were found to be ineffective for this coupling (16–26%) (entries 6–9), whereas the coupling partners stayed inert in the presence of AuBr3, CuI, and LiOTf (entries 10 and 11).
|
|
||
|---|---|---|
| Entry | Promoter (0.1 eq.) | Isolated yield (%) |
| 1 | PPh3AuNTf2 | 43 |
| 2 | TMSOTf | 91 |
| 3 | Bi(OTf)3 | 83 |
| 4 | In(OTf)3 | 67 |
| 5 | Cu(OTf)2 | 46 |
| 6 | Sc(OTf)3 | 17 |
| 7 | BF3OEt2 (1.2 eq.) | 21 |
| 8 | SnCl4 | 26 |
| 9 | PtCl2 | 16 |
| 10 | AuBr3 | Trace |
| 11 | CuI or LiOTf | No reaction |
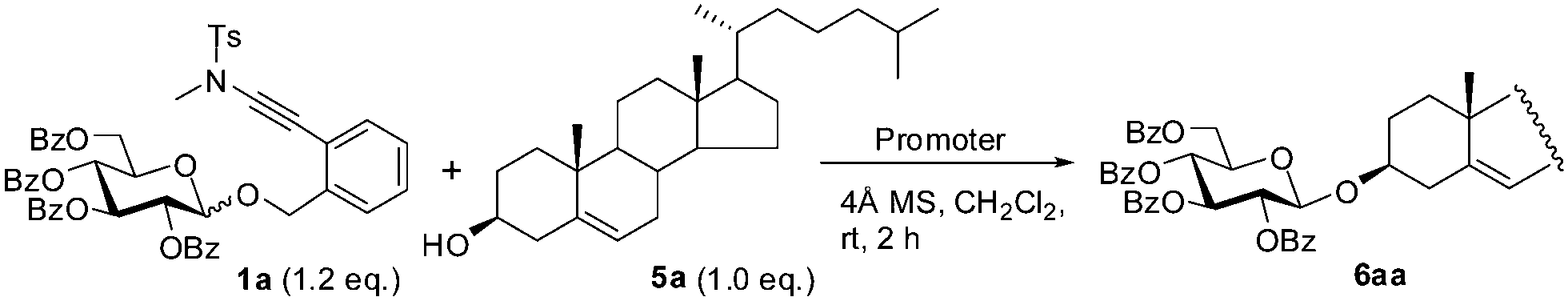
Next, we investigated briefly the scope of the TMSOTf-catalyzed glycosylation reaction with ortho-(methyltosylaminoethynyl)benzyl glycosides as donors (Table 2). Four representative glycosides 1a–1d and four alcohols 5a–5d were selected as coupling partners, and all the reactions were carried out under fixed conditions (0.1 eq. TMSOTf, 4 Å MS, CH2Cl2, rt, 2 h). The couplings of perbenzoyl-glucopyranoside 1a with all the four alcohols led to the coupled glycosides in excellent yields (>91%) and with complete β-selectivity (entries 1–4), testifying the participation of the neighbouring group in the glycosylation.1 Similarly, the couplings of 2,3,4-tri-O-benzoyl-L-rhamnopyranoside 1b with alcohols 5a–5d provided the corresponding α-L-rhamnosides in a fully stereocontrolled manner in high yields (88–95%; entries 5–8). As expected, the corresponding glycosylation reactions of perbenzylglucopyranoside 1c and 2-deoxy-glucopyranoside 1d, in the absence of a neighboring participating group, led to the coupled glycosides in high yields (83–99%), albeit in a pair of the α- and β-anomers (Table 2, entries 11–16).12,13 It was noted that the reactions with the hindered glucose-4-OH derivative 5c as the acceptor were devoid of the addition of alcohol onto the ynamide moiety, therefore the unglycosylated 5c could be fully recovered.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Donor | Acceptor | Product | Yielda [%] | α/βb |
| a Isolated yield. b The α/β ratio was determined by 1H NMR spectroscopic measurement. | |||||
| 1 | 1a | 5a | 6aa | 91 | β only |
| 2 | 5b | 6ab | 91 | β only | |
| 3 | 5c | 6ac | 98 | β only | |
| 4 | 5d | 6ad | 94 | β only | |
| 5 | 1b | 5a | 6ba | 90 | α only |
| 6 | 5b | 6bb | 95 | α only | |
| 7 | 5c | 6bc | 88 | α only | |
| 8 | 5d | 6bd | 91 | α only | |
| 9 | 1c | 5a | 6ca | 95 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 10 | 5b | 6cb | 93 | 1.5:1 |
|
| 11 | 5c | 6cc | 85 | 1.2:1 |
|
| 12 | 5d | 6cd | 89 | 1.2:1 |
|
| 13 | 1d | 5a | 6da | 93 | 1.8:1 |
| 14 | 5b | 6db | 99 | 2.5:1 |
|
| 15 | 5c | 6dc | 83 | 10:1 |
|
| 16 | 5d | 6dd | 89 | 3:1 |
|
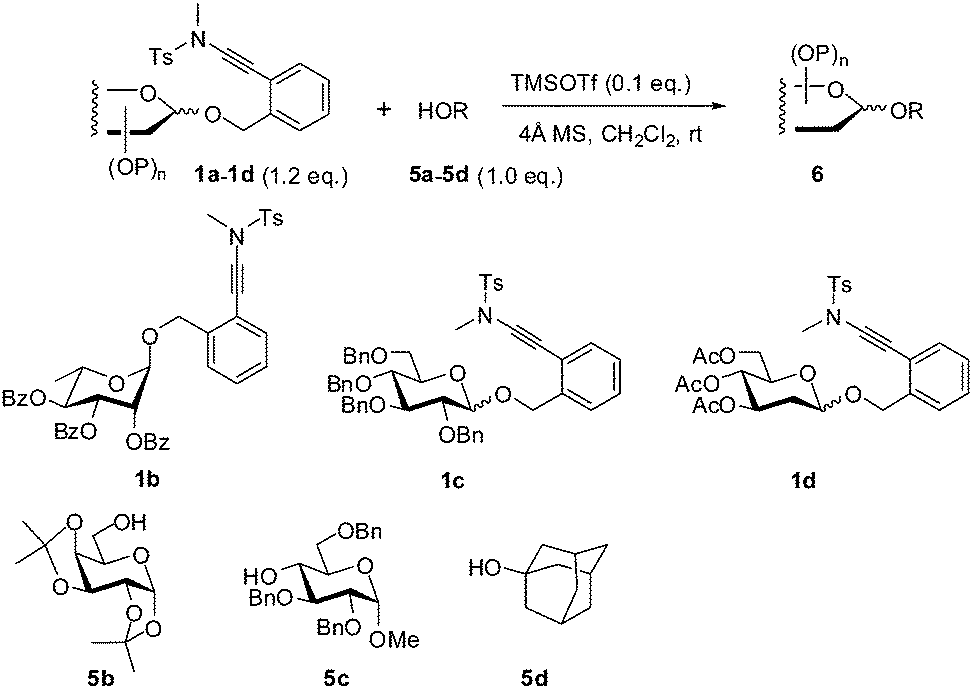
In fact, the addition of alcohol onto the ynamide moiety became a serious problem when the alcohol to be glycosylated is highly reactive. Thus, the condensation of 1a with 4-penten-1-ol 5e under the catalysis of TMSOTf delivered the coupled glycoside 6ae in only 62% yield, while ester 7, which was derived from the corresponding adduct during workup, was isolated in 34% yield (Scheme 2).11
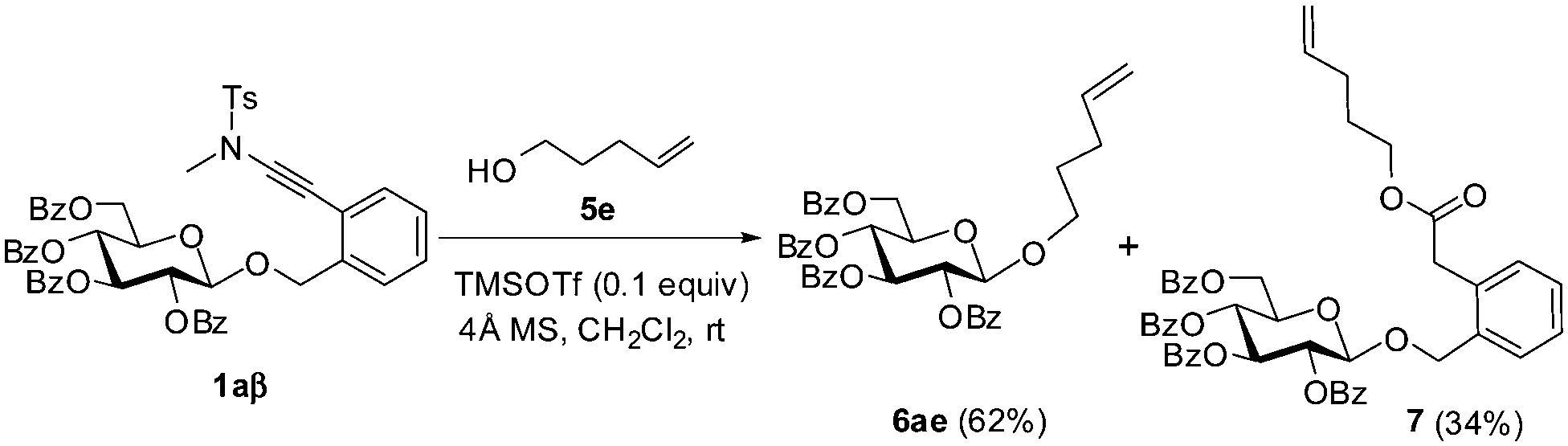 | ||
| Scheme 2 The coupling of ortho-(methyltosylaminoethynyl)benzyl glucopyranoside 1a with 4-penten-1-ol (5e). | ||
Based on these experimental findings and the nature of ynamides,9,11 a plausible mechanism for the present TMSOTf-catalyzed glycosylation reaction with ortho-(methyltosylaminoethynyl)benzyl glycosides as donors was proposed (Scheme 3). Thus, keteniminium cation B was generated from ortho-(methyltosylaminoethynyl)benzyl glycoside A in the presence of ROH and TMSOTf (wherein HOTf14 was produced in situ).15 An intramolecular nucleophilic addition of the anomeric oxygen onto the keteniminium led to sugar oxocarbenium ion C and 1H-isochromene D which was indeed characterized (path a). Sugar oxocarbenium ion C underwent glycosylation in the presence of ROH to provide glycoside E.1e Alternatively, keteniminium cation B could be attacked by the alcohol ROH, giving rise to alkoxy-substituted enamine intermediate F (path b). Hydrolysis of the enamine F during workup provided ester G.
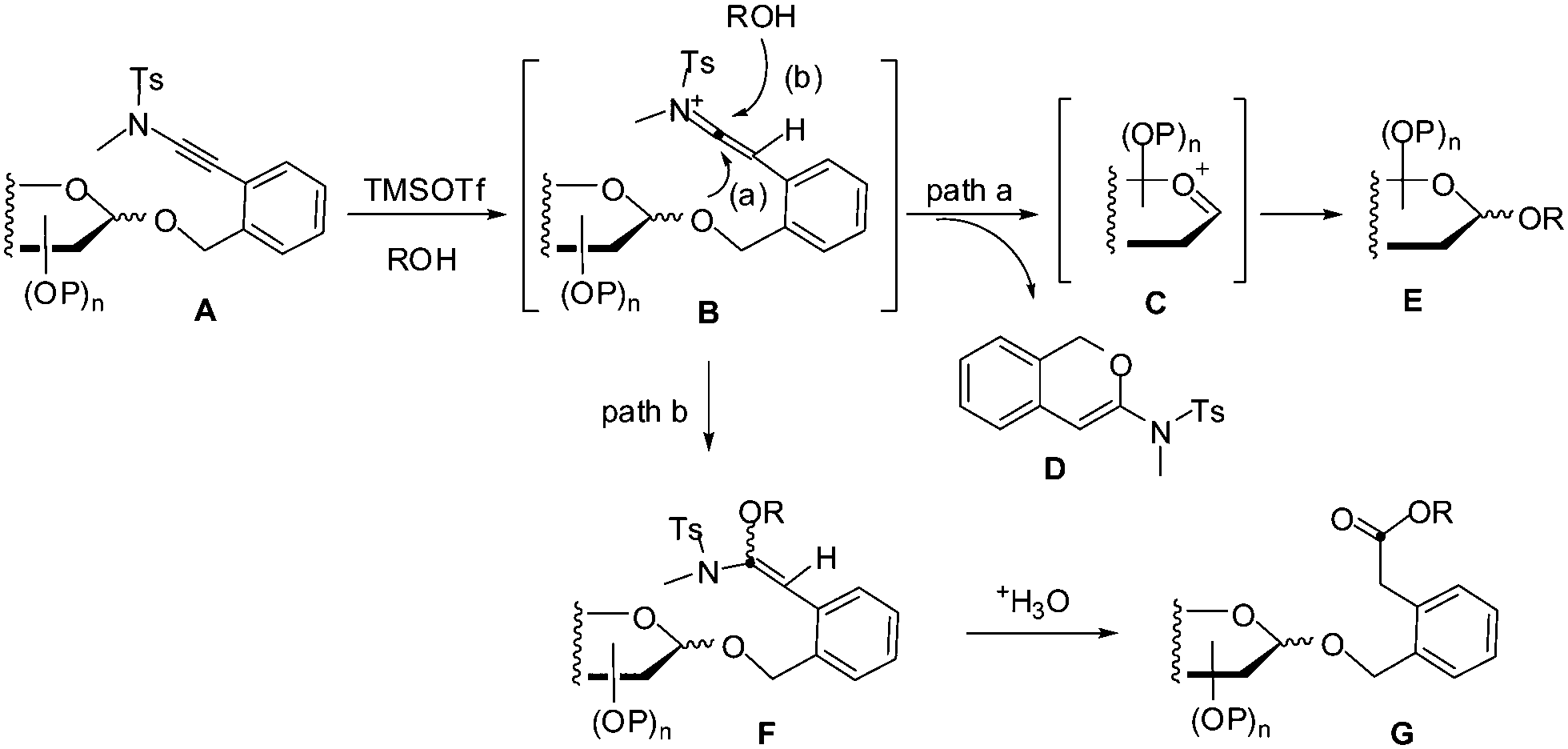 | ||
| Scheme 3 A plausible mechanism for the glycosylation with ortho-(methyltosylaminoethynyl)benzyl glycosides as donors under the catalysis of TMSOTf. | ||
Given the fact that the ortho-(methyltosylaminoethynyl)benzyl glycoside donors are readily prepared from the ortho-iodobenzyl glycosides, which are inactive in the glycosylation reactions of the former, these donors could be applied to the expeditious synthesis of oligosaccharides based on the ‘latent-active’ strategy.16 The previous donors applicable in the ‘latent-active’ synthesis of glycans include p-acetamidophenyl thioglycosides (vs. p-nitrophenyl thioglycosides),17n-pentenyl glycosides (vs. 4,5-dibromopentyl glycosides),18 vinyl glycoside (vs. 1-methyl-2-propenyl glycosides),19 2-(hydroxycarbonyl)benzyl glycosides (vs. 2-(benzyloxycarbonyl)benzyl glycosides),20 and S-benzimidazolyl glycosides (vs. N-anisoylated S-benzimidazolyl glycosides).21 To demonstrate the feasibility of applying the present glycosylation protocol to the ‘latent-active’ assembly of glycans, the ‘active’ ortho-(methyltosylaminoethynyl)benzyl glycoside 1a was coupled with the ‘latent’ ortho-iodobenzyl glucoside derivative 8 in the presence of TMSOTf (0.1 eq.) to provide β-(1→6)-disaccharide 9 (97%), which was then converted into the ‘active’ ortho-(methyltosylaminoethynyl)benzyl disaccharide 10via Sonogashira coupling with ynamide 4 (88%) (Scheme 4). Subsequent glycosylation of disaccharide 10 with cholesterol 5a or glucose-4-OH derivative 5c under similar glycosylation conditions furnished cholesterol 3-O-β-disaccharide 11 and β-trisaccharide 12 in 87% and 86% yields, respectively. In addition, glycosylation of disaccharide 10 with the ‘latent’ ortho-iodobenzyl glucoside acceptor 8 provided the ‘latent’ ortho-iodobenzyl trisaccharide 13 in 97% yield, which could be used for further elongation of the glycans via the iterative Sonogashira coupling/glycosylation sequence.
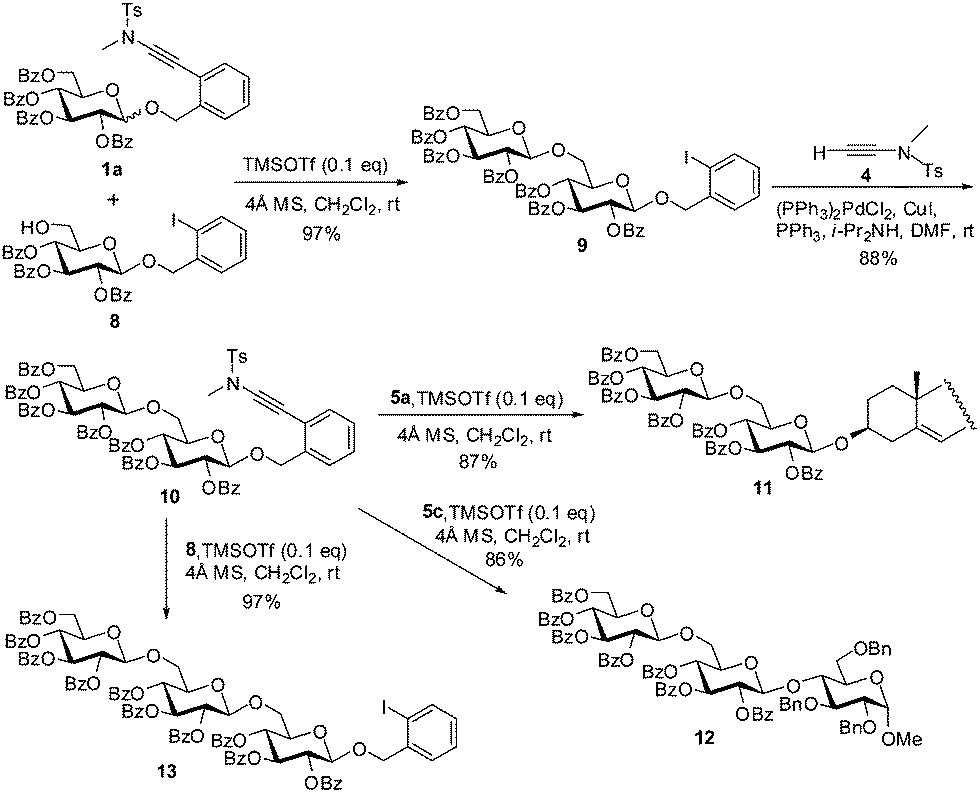 | ||
| Scheme 4 Assembly of oligosaccharides 11–13 by the ‘latent-active’ strategy using ortho-(methyltosylaminoethynyl)benzyl glycosides as donors and ortho-iodobenzyl glycosides as acceptors. | ||
In conclusion, ortho-(methyltosylaminoethynyl)benzyl glycosides have been disclosed as a new type of glycosyl donors under the catalysis of TMSOTf. These shelf-stable donors are readily prepared from the corresponding ortho-iodobenzyl glycosides via Sonogashira coupling with ynamide 4. The expeditious assembly of glycans via the ‘latent-active’ strategy using the present protocol has been demonstrated. These promising preliminary results shall warrant further elaboration and application of this new glycosylation method.
Financial support from the Ministry of Science and Technology of China (2012ZX09502-002), the National Natural Science Foundation of China (21432012), the Fundamental Research Funds for the Central Universities (WY1514052), and the Shanghai Pujiang Program (15PJ1401500) is gratefully acknowledged.
Notes and references
- (a) X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed; (b) M. J. McKay and H. M. Nguyen, ACS Catal., 2012, 2, 1563–1595 CrossRef CAS PubMed; (c) B. Yu, J. Sun and X. Yang, Acc. Chem. Res., 2012, 45, 1227–1236 CrossRef CAS PubMed; (d) S. S. Nigudkar and A. V. Demchenko, Chem. Sci., 2015, 6, 2687–2704 RSC; (e) L. Bohe and D. Crich, Carbohydr. Res., 2015, 403, 48–59 CrossRef CAS PubMed; (f) Y. Yang, X. Zhang and B. Yu, Nat. Prod. Rep., 2015 10.1039/C5NP00033E.
- (a) S. Hotha and S. Kashyap, J. Am. Chem. Soc., 2006, 128, 9620–9621 CrossRef CAS PubMed; (b) S. K. Mamidyala and M. G. Finn, J. Org. Chem., 2009, 74, 8417–8420 CrossRef CAS PubMed; (c) S. R. Vidadala, S. A. Thadke and S. Hotha, J. Org. Chem., 2009, 74, 9233–9236 CrossRef CAS PubMed; (d) S. R. Vidadala, G. Gayatri, G. N. Sastry and S. Hotha, Chem. Commun., 2011, 47, 9906–9908 RSC; (e) A. K. Kayastha and S. Hotha, Chem. Commun., 2012, 48, 7161–7163 RSC; (f) A. K. Kayastha and S. Hotha, Beilstein J. Org. Chem., 2013, 9, 2147–2155 CrossRef PubMed.
- (a) G. Sureshkumar and S. Hotha, Tetrahedron Lett., 2007, 48, 6564–6568 CrossRef CAS PubMed; (b) G. Sureshkumara and S. Hotha, Chem. Commun., 2008, 4282–4284 RSC; (c) A. Y. Shaikh, G. Sureshkumar, D. Pati, S. S. Guptab and S. Hotha, Org. Biomol. Chem., 2011, 9, 5951–5959 RSC; (d) S. A. Thadke, B. Mishra and S. Hotha, Org. Lett., 2013, 15, 2466–2469 CrossRef CAS PubMed; (e) S. A. Thadke, B. Mishra and S. Hotha, J. Org. Chem., 2014, 79, 7358–7371 CrossRef CAS PubMed; (f) B. V. Rao, S. Manmode and S. Hotha, J. Org. Chem., 2015, 80, 1499–1505 CrossRef CAS PubMed.
- H. Imagawa, A. Kinoshita, T. Fukuyama, H. Yamamoto and M. Nishizawa, Tetrahedron Lett., 2006, 47, 4729–4731 CrossRef CAS PubMed.
- S. R. Koppolu, R. Niddana and R. Balamurugan, Org. Biomol. Chem., 2015, 13, 5094–5097 CAS.
- (a) F. Yang, Q. Wang and B. Yu, Tetrahedron Lett., 2012, 53, 5231–5234 CrossRef CAS PubMed; (b) S. Adhikari, K. N. Baryal, D. Zhu, X. Li and J. Zhu, ACS Catal., 2013, 3, 57–60 CrossRef CAS.
- (a) Y. Li, Y. Yang and B. Yu, Tetrahedron Lett., 2008, 49, 3604–3608 CrossRef CAS PubMed; (b) Y. Li, X. Yang, Y. Liu, C. Zhu, Y. Yang and B. Yu, Chem. – Eur. J., 2010, 16, 1871–1882 CrossRef CAS PubMed; (c) Y. Zhu and B. Yu, Angew. Chem., Int. Ed., 2011, 50, 8329–8332 CrossRef CAS PubMed; (d) Y. Tang, J. Li, Y. Zhu, Y. Li and B. Yu, J. Am. Chem. Soc., 2013, 135, 18396–18405 CrossRef CAS PubMed; (e) Y. Zhu and B. Yu, Chem. – Eur. J., 2015, 21, 8771–8780 CrossRef CAS PubMed.
- (a) Y. Yang, Y. Li and B. Yu, J. Am. Chem. Soc., 2009, 131, 12076–12077 CrossRef CAS PubMed; (b) Y. Li and B. Yu, Chem. Commun., 2010, 46, 6060–6062 RSC; (c) W. Yang, J. Sun, W. Lu, Y. Li, L. Shan, W. Han, W.-D. Zhang and B. Yu, J. Org. Chem., 2010, 75, 6879–6888 CrossRef CAS PubMed; (d) Y. Ma, Z. Li, H. Shi, J. Zhang and B. Yu, J. Org. Chem., 2011, 76, 9748–9756 CrossRef CAS PubMed; (e) Q. Zhang, J. Sun, Y. Zhu, F. Zhang and B. Yu, Angew. Chem., Int. Ed., 2011, 50, 4933–4936 CrossRef CAS PubMed; (f) Y. Li, J. Sun and B. Yu, Org. Lett., 2011, 13, 5508–5511 CrossRef CAS PubMed; (g) J. Yu, J. Sun and B. Yu, Org. Lett., 2012, 14, 4022–4025 CrossRef CAS PubMed; (h) J. Zhang, H. Shi, Y. Ma and B. Yu, Chem. Commun., 2012, 48, 8679–8681 RSC; (i) J. Yu, J. Sun, Y. Niu, R. Li, J. Liao, F. Zhang and B. Yu, Chem. Sci., 2013, 4, 3899–3905 RSC; (j) G. Xiao and B. Yu, Chem. – Eur. J., 2013, 19, 7708–7712 CrossRef CAS PubMed; (k) S. Nie, W. Li and B. Yu, J. Am. Chem. Soc., 2014, 136, 4157–4160 CrossRef CAS PubMed; (l) J. Li and B. Yu, Angew. Chem., Int. Ed., 2015, 54, 6618–6621 CrossRef CAS PubMed; (m) X. Zhang, Y. Zhou, J. Zuo and B. Yu, Nat. Commun., 2015, 6, 5879, DOI:10.1038/ncomms6879.
- (a) C. A. Zificsak, J. A. Mulder, R. P. Hsung, C. Rameshkumar and L.-L. Wei, Tetrahedron, 2001, 57, 7575–7606 CrossRef CAS; (b) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840–2859 CrossRef CAS PubMed.
- (a) Y.-P. Wang and R. L. Danheis, Tetrahedron Lett., 2011, 52, 2111–2114 CrossRef CAS PubMed; (b) M. R. Tracey, Y. Zhang, M. O. Frederick, J. A. Mulder and R. P. Hsung, Org. Lett., 2011, 13, 5508–5511 CrossRef PubMed.
- (a) M. C. B. Jaimes, V. Weingand, F. Rominger and A. S. K. Hashmi, Chem. – Eur. J., 2013, 19, 12504–12511 CrossRef PubMed; (b) Y. Kong, K. Jiang, J. Cao, L. Fu, L. Yu, G. Lai, Y. Cui, Z. Hu and G. Wang, Org. Lett., 2013, 15, 422–425 CrossRef CAS PubMed; (c) T. Okitsu, K. Nakata, K. Nishigaki, N. Michioka, M. Karatani and A. Wada, J. Org. Chem., 2014, 79, 5914–5920 CrossRef CAS PubMed; (d) D. Fujino, H. Yorimitsu and A. Osuka, J. Am. Chem. Soc., 2014, 136, 6255–6258 CrossRef CAS PubMed; (e) S. Kramer, K. Dooleweerdt, A. T. Lindhardt, M. Rottländer and T. Skrydstrup, Org. Lett., 2009, 11, 4208–4211 CrossRef CAS PubMed.
- D. Hou and T. L. Lowary, Carbohydr. Res., 2009, 344, 1911–1940 CrossRef CAS PubMed.
- E. Juaristi and G. Cuevas, The Anomeric Effect, CRC Press, 1994, pp. 183–194 Search PubMed.
- T. T. Dang, F. Boeck and L. Hintermann, J. Org. Chem., 2011, 76, 9353–9361 CrossRef CAS PubMed.
- Y. Zhang, R. P. Hsung, X. Zhang, J. Huang, B. W. Slafer and A. Davis, Org. Lett., 2005, 7, 1047–1050 CrossRef CAS PubMed.
- (a) T. C. Shiao and R. Roy, Top. Curr. Chem., 2011, 301, 69–108 CrossRef CAS; (b) J. T. Smoot and A. V. Demchenko, Adv. Carbohydr. Chem. Biochem., 2009, 62, 161–250 CrossRef CAS; (c) G. J. Boons, Tetrahedron, 1996, 52, 1095–1121 CrossRef CAS.
- (a) R. Roy, F. O. Andersson and M. Letellier, Tetrahedron Lett., 1992, 33, 6053–6056 CrossRef CAS; (b) L. Huang, Z. Wang and X. Huang, Chem. Commun., 2004, 1960–1961 RSC.
- B. Fraser-Reid, U. E. Udodong, Z. F. Wu, H. Ottosson, J. R. Merritt, C. S. Rao, C. Roberts and R. Madsen, Synlett, 1992, 927–942 CrossRef CAS.
- (a) G.-J. Boons and S. Isles, Tetrahedron Lett., 1994, 35, 3593–3596 CrossRef CAS; (b) P. Wang, P. Haldar, Y. Wang and H. Hu, J. Org. Chem., 2007, 72, 5870–5873 CrossRef CAS PubMed.
- K. S. Kim, J. H. Kim, Y. J. Lee, Y. J. Lee and J. Park, J. Am. Chem. Soc., 2001, 123, 8477–8481 CrossRef CAS PubMed.
- S. J. Hasty, M. A. Kleine and A. V. Demchenko, Angew. Chem., Int. Ed., 2011, 50, 4197–4201 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc05651a |
| This journal is © The Royal Society of Chemistry 2015 |