
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGaining insight into the catalysis by GH20 lacto-N-biosidase using small molecule inhibitors and structural analysis†
Mitchell
Hattie
a,
Tasuku
Ito
b,
Aleksandra W.
Debowski
ac,
Takatoshi
Arakawa
d,
Takane
Katayama
e,
Kenji
Yamamoto
f,
Shinya
Fushinobu
d and
Keith A.
Stubbs
*a
aSchool of Chemistry and Biochemistry, The University of Western Australia, Crawley, WA 6009, Australia. E-mail: keith.stubbs@uwa.edu.au
bNational Food Research Institute, National Agriculture and Food Research Organization, Tsukuba, Ibaraki 305-8642, Japan
cSchool of Pathology and Laboratory Medicine, The University of Western Australia, Crawley, WA 6009, Australia
dDepartment of Biotechnology, The University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan
eGraduate School of Biostudies, Kyoto University, Kyoto 606-8502, Japan
fResearch Institute for Bioresources and Biotechnology, Ishikawa Prefectural University, Nonoichi, Ishikawa 921-8836, Japan
First published on 27th August 2015
Abstract
The synthesis of potent inhibitors for lacto-N-biosidases and X-ray structural characterization of these compounds in complex with BbLNBase is described.
The role that the microbiome plays in human health and disease is being shown to be extremely important and as such is receiving considerable attention.1 Bacteria of the genus Bifidobacterium are especially critical to the health of the GI tract as they constitute a large proportion of the GI microbiome2 and they have been shown to be important in influencing the distribution of other GI microbiota.3 Colonization of the GI tract by these bacteria occurs soon after birth and it is believed that they play a beneficial role in stimulating the immune response, preventing colonization of pathogenic bacteria and suppressing inflammatory responses.4,5
To establish and maintain colonization bifidobacteria express a wide range of carbohydrate-processing enzymes2 which allows them to utilize carbohydrates that are not digestible by the host, or other microbes. This trait offers a competitive advantage especially in breast-fed infants.6 One specific class of carbohydrates that is acted upon by bifidobacterial enzymes are the human milk oligosaccharides (HMOs),7 that include over 130 different glycans and are found in concentrations of up to 20 g per litre in human milk.8,9 Due to the importance of these compounds for the life of bifidobacteria,10 unique biochemical pathways have evolved in these bacteria to breakdown these compounds. One such pathway is termed the lacto-N-biose (LNB) pathway11,12 which allows for metabolic utilization of LNB (Gal-(β1,3)-GlcNAc),13 a common structural motif found in HMOs.9,14 Consequently, an enzyme important to the LNB pathway is lacto-N-biosidase (LNBase), a β-N-acetyl-hexosaminidase that liberates LNB from HMOs.
LNBases are classified currently as members of family 20 of the glycoside hydrolases (GHs)‡ and like other members of this family it has been shown to use a two-step catalytic mechanism involving substrate-assisted catalysis that forms a transient oxazoline or oxazolinium ion intermediate (Fig. 1A).15,16 Much of the insight into the active site architectures and catalytic mechanisms of GH20 glycosidases and several other glycosidase families has been made through the synthesis of inhibitors of the respective enzyme being studied. In respect to LNBase, the disaccharide 1 has been prepared,15 which is based on the known inhibitor NAG-thiazoline,17 a potent inhibitor of other GH20 exo-β-N-acetyl-hexosaminidases which only cleave monosaccharide GlcNAc (or GalNAc) residues from glycoconjugates. Indeed, the disaccharide 1 was shown to be a potent inhibitor and aided in the confirmation of the catalytic mechanism of LNBases.15,16
 | ||
| Fig. 1 (A) Reaction catalysed by BbLNBase with the oxazoline intermediate and putative transition state shown (R1 = β-D-galactopyranose). (B) Inhibitors of other β-N-acetylhexosamindases as well as current and presented inhibitors of LNBase. | ||
Due to the considerable interest in exo-β-N-acetyl-hexosaminidases from GH20 and other GH families, numerous other potent inhibitors have also been synthesized with the most well known of these being the hydroximolactone-based compounds LOGNAc and PUGNAc,18 the iminosugars NHAcDNJ,19 and NHAcCAS and an isomer of the latter compound NHAc-australine20 (Fig. 1B). The inhibitory properties of these compounds towards exo-β-N-acetyl-hexosaminidases is thought to come about through the way each of them mimic, either through shape or charge, the putative transition state of the pyranose ring during catalysis (Fig. 1A).21 As LNBases are important enzymes in the degradation of HMOs by bifidobacteria and that thiazoline-based inhibitors suffer from instability in solution,22 the development of a repertoire of suitable tools is necessary to study the roles that this enzyme plays in bacterial growth and establishment within the GI tract. Thus we set about preparing compounds 2–6 which are compounds tailor-made to be potent inhibitors of LNBase.
Compounds 2 and 3 were prepared starting from the alcohol 723 (Scheme 1). TMSOTf-promoted glycosylation with the trichloroacetimidate 8, gave the disaccharide 9 in good yield. Removal of the benzylidene protecting group gave the diol 10, which was then converted to the acetate 11. Of note here is that 10 is a key intermediate to access not only 2 and 3 but also the iminosugar 4. Treatment of 11 under acetolysis conditions followed by selective removal of the presumed anomeric acetate gave the hemiacetal 12. This material was then activated with hydroxylamine hydrochloride which yielded the presumed mixture of oximes which was in turn converted to the hydroximolactone 13 in excellent overall yield. For the preparation of 2, acetylation of 13 gave 14, and subsequent conversion of the azide to an acetamido group using Pd(OH)2-mediated hydrogenolysis gave 15. Global deprotection furnished the desired compound 2 in good overall yield. For 3, the hydroximolactone 13 was converted to the carbamate 16 and using similar conditions as for the preparation of 15 and 2, compounds 17 and 3 were prepared respectively.
 | ||
Scheme 1 (a) TMSOTf, 4A sieves, CH2Cl2; (b) CH3COOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (4:1); (c) Ac2O, pyr.; (d) i. Ac2O, H2SO4; ii. Aq. MeNH2, THF; (e) i. NH2OH·HCl, pyr.; ii. DBU, NCS, CH2Cl2; (f) Ac2O, pyr. CH2Cl2; (g) Pd(OH)2/C, H2, Ac2O, EtOH; (h) NH3, MeOH; (i) PhNCO, Et3N, THF; (j) i. TsCl, pyr., CH2Cl2; ii. NaI, DMF; iii. Ac2O, pyr.; (k) DBU, THF; (l) PBu3, Ac2O, pyr, THF, H2O; (m) i. mCPBA, BnOH, CH2Cl2; ii. NaOMe, MeOH; iii. NH4HCOO, Pd(OH)2/C, H2, MeOH, H2O; (n) i. CH3COOH:H2O (4:1); ii. Ac2O, pyr.; (o) Pd/C, H2, MeOH; (p) i. MsCl, pyr.; ii. NaN3, DMSO; (q) i. Pd/C, H2, PhMe; ii. Ac2O, pyr; (r) NaOMe, MeOH. :H2O (4:1); (c) Ac2O, pyr.; (d) i. Ac2O, H2SO4; ii. Aq. MeNH2, THF; (e) i. NH2OH·HCl, pyr.; ii. DBU, NCS, CH2Cl2; (f) Ac2O, pyr. CH2Cl2; (g) Pd(OH)2/C, H2, Ac2O, EtOH; (h) NH3, MeOH; (i) PhNCO, Et3N, THF; (j) i. TsCl, pyr., CH2Cl2; ii. NaI, DMF; iii. Ac2O, pyr.; (k) DBU, THF; (l) PBu3, Ac2O, pyr, THF, H2O; (m) i. mCPBA, BnOH, CH2Cl2; ii. NaOMe, MeOH; iii. NH4HCOO, Pd(OH)2/C, H2, MeOH, H2O; (n) i. CH3COOH:H2O (4:1); ii. Ac2O, pyr.; (o) Pd/C, H2, MeOH; (p) i. MsCl, pyr.; ii. NaN3, DMSO; (q) i. Pd/C, H2, PhMe; ii. Ac2O, pyr; (r) NaOMe, MeOH. | ||
The iminosugar 4, was synthesized from 10 starting with a one-pot activation at O-6, and in situ acetylation to give the iodide 18 in excellent yield over three steps. Elimination across C-5/6 was achieved using DBU to give the desired alkene 19. Treatment of the alkene 19 using Staudinger conditions followed by acetylation gave the amide 20. Oxidation of 20 with mCPBA, followed by deprotection provided the presumed intermediate ulososide which when put under reductive amination conditions in the presence of ammonium formate and hydrogen gratifyingly gave 4, exclusively as the D-isomer, consistent with previous observations for similar transformations.24,25
The synthesis of the iminosugars 5 and 6 started from the carbamate 21.26 Again, TMSOTf-promoted glycosylation with the trichloroacetimidate 8, gave the disaccharide 22 in good yield. Removal of the isopropylidene protecting group and in situ acetylation gave 23. Removal of the Cbz-protecting group yielded the alcohol 24, which is the critical divergence point to the synthesis of 5 and 6. Activation of the alcohol on 24 as a presumed mesylate, followed by treatment with sodium azide20 gave the two azides 25 and 26. For 5, hydrogenolysis of 25 followed by in situ acetylation gave 27, which after removal of the acetyl protecting groups gave the desired disaccharide 5. Similar methodologies were used for converting crude 26, through 28, to 6.
With the synthesized set of inhibitors now in hand, we evaluated them against a representative GH20 LNBase found in Bifidobacterium bifidum (BbLNBase) and found them all to be potent competitive inhibitors of this enzyme (Table 1). These results demonstrate that these inhibitor scaffolds that have been used previously to inhibit exo-β-N-acetyl-hexosaminidases are also useful in inhibiting the disaccharide releasing LNBase-type enzymes. In terms of comparing the potency of the inhibitors 2–6 to their monosaccharide derivatives, all the compounds are in good agreement. Typically, PUGNAc is a very potent inhibitor of GH20 and GH84 enzymes27,28 with LOGNAc being less potent.29 The iminosugars NHAcDNJ and NHAcCAS also have good potency towards enzymes from GH2030 and GH84.25,31 NHAc-australine has to date, not been assessed as an inhibitor of exo-β-N-acetyl-hexosaminidases.
| Compound | K i (μM) |
|---|---|
| 1 | 0.125 ± 0.00815 |
| 2 | 7.7 ± 0.1 |
| 3 | 0.091 ± 0.003 |
| 4 | 0.88 ± 0.012 |
| 5 | 0.52 ± 0.007 |
| 6 | 52 ± 2 |
To gain a more detailed understanding of the molecular basis for the inhibition of BbLNBase co-crystallization trials with 2–6 were attempted. We finally determined high resolution (up to 1.60 Å) crystal structures of BbLNBase in complex with compounds 2, 4–6 (Fig. 2) with clear electron densities. Of note is that this is also the first report of an X-ray structure of an exo-β-N-acetyl-hexosaminidase in complex with a NHAc-australine-based compound. All of the hydroxyl groups of the inhibitors 2, 4–6 form hydrogen bonds with the surrounding amino acids. Previously it has been established that the amino acid D467 plays a crucial role in recognizing the pyranose ring of LNB and 1 at the −1 subsite, forming bidentate hydrogen bonds with the C4 and C6 hydroxyl groups.16 In concert with these observations, the bidendate hydrogen bonds from D467 are present in all complex structures, even in 5 and 6, which have an unusual bicyclic group in the −1 subsite. The hydroxyl group present at the C1 position of the iminosugar in 5 is located at the most appropriate position to form a hydrogen bond whereas the corresponding hydroxyl group in 6 is not ideally positioned. This difference in the geometry of this hydrogen bond is likely a source of the difference in the Ki values obtained for 5 and 6.
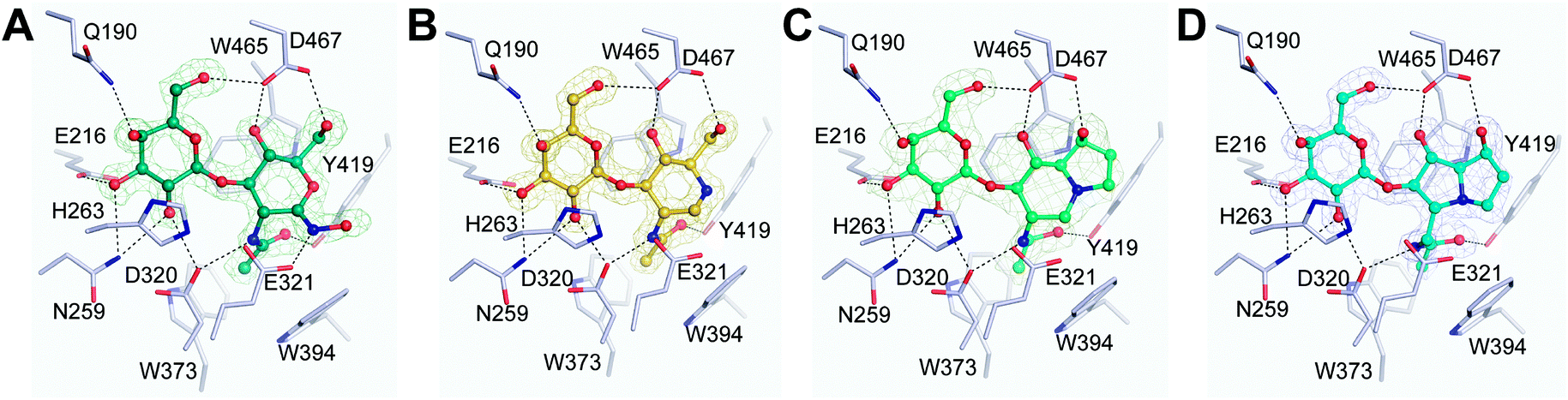 | ||
| Fig. 2 Active site structures of BbLNBase in complex with 2 (A), 4 (B), 5 (C), and 6 (D). |Fo| − |Fc| omit electron density maps (mesh, 1.5σ) and hydrogen bonds (dashed lines) are shown with a hydrogen bond cut off distance of 3.2 Å used. | ||
For the hydroximolactone-based compound 2, similar features are observed with an additional hydrogen bond also being formed between the nitrogen atom of the hydroximo group and E321 (catalytic acid/base). Due to lack of success in obtaining BbLNBase in complex with 3, a docking analysis was undertaken in an effort to gain insight into the molecular basis for inhibition. Using Autodock Vina,32 a good match was obtained for the binding of 3 with BbLNBase, using the protein structure of BbLNBase–2 complex as a receptor, with the affinity determined to be −8.7 kcal mol−1 which is in good agreement with the Ki (see figure in ESI†). In addition to similar binding features observed for 2, 4–6 the docking analysis also revealed a potential extra hydrogen bond between the hydroximolactone oxygen of 3 and Y427. Interestingly the docking analysis also revealed that the phenyl ring is positioned in a hydrophobic pocket of BbLNBase surrounded by A424 and V426 which lies ahead of the hydrophobic cage commonly found in GH20 exo-β-N-acetyl-hexosaminidases,33 and this likely also adds to the increased potency of 3versus2.
One method that can lead to inhibition of glycosidases is to mimic the conformation of the pyranose ring at the intermediate and transition states of catalysis (Fig. 1A).21 Thus we analysed the conformations of the pyranose ring containing inhibitors 2, 4 and 5 in detail using the Cremer–Pople system which is used to determine the conformation of six-membered rings (see table in ESI†).34 The inhibitor 2 is in a typical 4E conformation (244° < φ < 247° and 59° < θ < 65°), similar to what is observed for the binding of corresponding inhibitors with GH2035 and GH84 enzymes.36 This conformation is also close to the putative transition states in the proposed conformational itinerary of the pyranose ring for BbLNBase16 and other enzymes that utilize a substrate-assisted catalytic mechanism.27,33,37 In contrast, the iminosugars 4 and 5 adopt a 1,4B conformation (234° < φ < 246° and 75° < θ < 85°). Based on these values it seems that both these compounds mimic somewhat the conformation that has been observed for Michaelis-like complex structures of GH20 exo-β-N-acetyl-hexosaminidases.38,39 These results give further credence to the proposed reaction pathway of BbLNBase: 1,4B (Michaelis complex)–4E (transition state)–4C1 (oxazoline intermediate)–4E (transition state)–4E (product complex).16
In conclusion the study of LNBases is critical to understanding how Bifidobacteria degrade HMOs and thus occupy a niche in the GI tract. The inhibitors prepared here are all potent inhibitors of BbLNBase and, through structural analysis, reasons for their potency are presented. Further detailed structural analysis of BbLNBase in complex with inhibitors synthesized by rational design will facilitate the development of more potent and stable inhibitors of LNBases. Additionally, these compounds will also prove useful for studying the roles that this enzyme plays in the bifidobacteria life cycle, HMO degradation and other biological pathways.40
The authors wish to thank the Centre for Microscopy, Characterisation and Analysis, The University of Western Australia, which is supported by University, State and Federal Government funding. KAS also thanks the Australian Research Council for funding. MH is supported by an Australian Postgraduate Award from the University of Western Australia and a Jean Rogerson Postgraduate Scholarship. AWD thanks the National Health and Medical Research Council for funding (APP1073250). SF thanks the staff of the Photon Factory for the X-ray data collection. A part of this work was supported by Platform for Drug Discovery, Informatics, and Structural Life Science funded by the Ministry of Education, Culture, Sports, Science and Technology, Japan, and by Science and Technology Research Promotion Program for Agriculture, Forestry, Fisheries and Food Industry.
Notes and references
- I. Cho and M. J. Blaser, Nat. Rev. Genet., 2012, 13, 260–270 CAS.
- T. Katayama, K. Fujita and K. Yamamoto, J. Biosci. Bioeng., 2005, 99, 457–465 CrossRef CAS PubMed.
- F. Turroni, F. Bottacini, E. Foroni, I. Mulder, J. H. Kim, A. Zomer, B. Sánchez, A. Bidossi, A. Ferrarini, V. Giubellini and M. Delledonne, et al. , Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19514–19519 CrossRef CAS PubMed.
- N. Iwabuchi, N. Takahashi, J. Z. Xiao, K. Miyaji and K. Iwatsuki, Microbiol. Immunol., 2007, 51, 649–660 CrossRef CAS PubMed.
- B. Sánchez, L. Ruiz, M. Gueimonde, P. Ruas-Madiedo and A. Margolles, Pharmacol. Res., 2013, 69, 127–136 CrossRef PubMed.
- K. Pokusaeva, G. F. Fitzgerald and D. van Sinderen, Genes Nutr., 2011, 6, 285–306 CrossRef CAS PubMed.
- A. Marcobal and J. Sonnenburg, Clin. Microbiol. Infect., 2012, 18, 12–15 CrossRef CAS PubMed.
- C. Kunz, S. Rudloff, W. Baier, N. Klein and S. Strobel, Annu. Rev. Nutr., 2000, 20, 699–722 CrossRef CAS PubMed.
- T. Urashima, T. Saito, T. Nakamura and M. Messer, Glycoconjugate J., 2001, 18, 357–371 CrossRef CAS.
- R. G. LoCascio, M. R. Ninonuevo, S. L. Freeman, D. A. Sela, R. Grimm, C. B. Lebrilla, D. A. Mills and J. B. German, J. Agric. Food Chem., 2007, 55, 8914–8919 CrossRef CAS PubMed.
- M. Kitaoka, J. Tian and M. Nishimoto, Appl. Environ. Microbiol., 2005, 71, 3158–3162 CrossRef CAS PubMed.
- S. Asakuma, E. Hatakeyama, T. Urashima, E. Yoshida, T. Katayama, K. Yamamoto, H. Kumagai, H. Ashida, J. Hirose and M. Kitaoka, J. Biol. Chem., 2011, 286, 34583–34592 CrossRef CAS PubMed.
- J. Wada, T. Ando, M. Kiyohara, H. Ashida, M. Kitaoka, M. Yamaguchi, H. Kumagai, T. Katayama and K. Yamamoto, Appl. Environ. Microbiol., 2008, 74, 3996–4004 CrossRef CAS PubMed.
- S. Asakuma, T. Urashima, M. Akahori, H. Obayashi, T. Nakamura, K. Kimura, Y. Watanabe, I. Arai and Y. Sanai, Eur. J. Clin. Nutr., 2008, 62, 488–494 CrossRef CAS PubMed.
- M. Hattie, A. W. Debowski and K. A. Stubbs, ChemBioChem, 2012, 13, 1128–1131 CrossRef CAS PubMed.
- T. Ito, T. Katayama, M. Hattie, H. Sakurama, J. Wada, R. Suzuki, H. Ashida, T. Wakagi, K. Yamamoto, K. A. Stubbs and S. Fushinobu, J. Biol. Chem., 2013, 288, 11795–11806 CrossRef CAS PubMed.
- S. Knapp, D. Vocadlo, Z. N. Gao, B. Kirk, J. P. Lou and S. G. Withers, J. Am. Chem. Soc., 1996, 118, 6804–6805 CrossRef CAS.
- D. Beer, J. L. Maloisel, D. M. Rast and A. Vasella, Helv. Chim. Acta, 1990, 73, 1918–1922 CrossRef CAS PubMed.
- E. Kappes and G. Legler, J. Carbohydr. Chem., 1989, 8, 371–388 CrossRef CAS PubMed.
- R. H. Furneaux, G. J. Gainsford, J. M. Mason and P. C. Tyler, Tetrahedron, 1994, 50, 2131–2160 CrossRef CAS.
- D. J. Vocadlo and G. J. Davies, Curr. Opin. Chem. Biol., 2008, 12, 539–555 CrossRef CAS PubMed.
- S. A. Yuzwa, M. S. Macauley, J. E. Heinonen, X. Shan, R. J. Dennis, Y. He, G. E. Whitworth, K. A. Stubbs, E. J. McEachern, G. J. Davies and D. J. Vocadlo, Nat. Chem. Biol., 2008, 4, 483–490 CrossRef CAS PubMed.
- B. K. S. Yeung, D. C. Hill, M. Janicka and P. A. Petillo, Org. Lett., 2000, 2, 1279–1282 CrossRef CAS.
- A. J. Steiner, G. Schitter, A. E. Stutz, T. M. Wrondigg, C. A. Tarling, S. G. Withers, D. J. Mahuran and M. J. Tropak, Tetrahedron: Asymmetry, 2009, 20, 832–835 CrossRef CAS PubMed.
- K. A. Stubbs, J. P. Bacik, G. E. Perley-Robertson, G. E. Whitworth, T. M. Gloster, D. J. Vocadlo and B. L. Mark, ChemBioChem, 2013, 14, 1973–1981 CrossRef CAS PubMed.
- P. S. Liu, M. S. Kang and P. S. Sunkara, Tetrahedron Lett., 1991, 32, 719–720 CrossRef CAS.
- M. S. Macauley, G. E. Whitworth, A. W. Debowski, D. Chin and D. J. Vocadlo, J. Biol. Chem., 2005, 280, 25313–25322 CrossRef CAS PubMed.
- K. A. Stubbs, M. S. Macauley and D. J. Vocadlo, Angew. Chem., Int. Ed., 2009, 48, 1300–1303 CrossRef CAS PubMed.
- K. A. Stubbs, M. Balcewich, B. L. Mark and D. J. Vocadlo, J. Biol. Chem., 2007, 282, 21382–21391 CrossRef CAS PubMed.
- M. B. Tropak, S. P. Reid, M. Guiral, S. G. Withers and D. Mahuran, J. Biol. Chem., 2004, 279, 13478–13487 CrossRef CAS PubMed.
- M. S. Macauley, Y. He, T. M. Gloster, K. A. Stubbs, G. J. Davies and D. J. Vocadlo, Chem. Biol., 2010, 17, 937–948 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- B. L. Mark, D. J. Vocadlo, S. Knapp, B. L. Triggs-Raine, S. G. Withers and M. N. James, J. Biol. Chem., 2001, 276, 10330–10337 CrossRef CAS PubMed.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- T. Liu, H. Zhang, H. Liu, L. Chen, X. Shen and Q. Yang, Biochem. J., 2011, 438, 467–474 CrossRef CAS PubMed.
- F. V. Rao, H. C. Dorfmueller, F. Villa, M. Allwood, I. M. Eggleston and D. M. van Aalten, EMBO J., 2006, 25, 1569–1578 CrossRef CAS PubMed.
- Y. He, M. S. Macauley, K. A. Stubbs, D. J. Vocadlo and G. J. Davies, J. Am. Chem. Soc., 2010, 132, 1807–1809 CrossRef CAS PubMed.
- I. Tews, A. Perrakis, A. Oppenheim, Z. Dauter, K. S. Wilson and C. E. Vorgias, Nat. Struct. Biol., 1996, 3, 638–648 CrossRef CAS PubMed.
- G. Prag, Y. Papanikolau, G. Tavlas, C. E. Vorgias, K. Petratos and A. B. Oppenheim, J. Mol. Biol., 2000, 300, 611–617 CrossRef CAS PubMed.
- A. Gotoh, T. Katoh, Y. Sugiyama, S. Kurihara, Y. Honda, H. Sakurama, T. Kambe, H. Ashida, M. Kitaoka, K. Yamamoto and T. Katayama, Carbohydr. Res., 2015, 408, 18–24 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc05494j |
| ‡ A LNBase from B. longum is currently non-classified. |
| This journal is © The Royal Society of Chemistry 2015 |