
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective aerobic oxidative Heck reactions with electronically unbiased alkenes: efficient access to α-alkyl vinylarenes†
Changwu
Zheng
and
Shannon S.
Stahl
*
Department of Chemistry, University of Wisconsin-Madison, 1101 University Avenue, Madison, WI 53706, USA. E-mail: stahl@chem.wisc.edu
First published on 3rd July 2015
Abstract
Branched-selective oxidative Heck coupling reactions have been developed between arylboronic acids and electronically unbiased terminal alkenes. The reactions exhibit high catalyst-controlled regioselectivity favoring the less common branched isomer. The reactions employ a catalyst composed of Pd(TFA)2/dmphen (TFA = trifluoroacetate, dmphen = 2,9-dimethyl-1,10-phenanthroline) and proceed efficiently at 45–60 °C under 1 atm of O2 without requiring other additives. A broad array of functional groups, including aryl halide, allyl silane and carboxylic acids are tolerated.
Mizoroki–Heck coupling reactions have found widespread use in organic synthesis1 and are among the most versatile methods for selective functionalization of vinylic C–H bonds.2 The ability to use unfunctionalized alkenes as substrates rather than vinyl halides represents a significant advantage over other cross-coupling methods for the synthesis of substituted alkenes. Several challenges, however, limit the scope of these reactions and prevent the advantages from being fully realized. Most precedents achieve regioselectivity by using substrates that have an intrinsic electronic bias (Scheme 1). Electron-deficient alkenes such as acrylates and styrenes favor coupling at the terminal position to give the linear products,3 while electron-rich vinyl ethers and vinyl amides favor coupling adjacent to the heteroatom to give the branched products.4 When electronically unbiased alkenes are used, the reaction often undergoes competitive reactions to afford different regioisomers with low selectivity, typically favoring the linear regioisomer. These issues were illustrated by Heck in 1974 in the reaction of bromobenzene with n-hexene, which under commonly used conditions afforded the linear phenylhexene as the major product, together with three other isomeric products.5
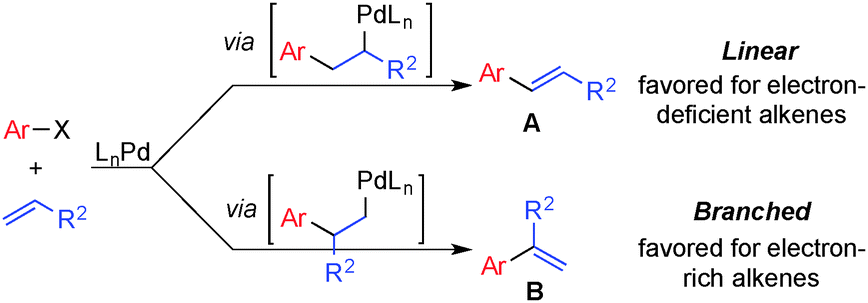 | ||
| Scheme 1 Regioselectivity in Pd-catalyzed Heck coupling reactions. | ||
Recent developments have begun to address these limitations through the identification of appropriate substrate partners, reaction conditions and catalyst systems. For example, White6a,b and co-workers achieved substrate-controlled linear-selective product formation with electronically unbiased alkenes via the use of chelating functional groups in the alkene substrate. Sigman6c,det al. identified appropriate catalyst systems that could achieve linear-selective coupling of simple alkenes with arylboronic acids or aryl diazonium salts.
Branched-selective Heck coupling typically requires electron-rich substrates, as noted above,4,7 but recent advances have led to catalysts capable of achieving branched selectivity with unbiased alkenes. Specifically, Zhou and co-workers reported Pd0-catalyzed Heck-coupling reactions between aliphatic olefins and aryl triflates using a bulky bisphosphine “dnpf” ligand [dnpf = 1,1′-bis[di(1-naphthyl)phosphino]-ferrocene].8 Jamison and co-workers reported Ni-catalyzed Heck-type coupling reactions of aryl sulfonates and related substrates with terminal olefins that achieve high levels of branched selectivity.9,10 Finally, in connection with our interest in Pd-catalyzed aerobic oxidative coupling reactions,11 we recently developed a method for oxidative Heck coupling of vinylboronic acids and electronically unbiased alkenes that afford branched-selective 1,3-disubstituted conjugated dienes.12 Here, we describe aerobic oxidative Heck reactions between arylboronic acids and electronically unbiased alkenes that proceed with high catalyst-controlled selectivity for the branched isomer. The sterically encumbered 2,9-dimethyl-1,10-phenanthroline (dmphen) ligand is crucial to control of the regioselectivity (Scheme 2).13 The resulting α-alkyl vinylarenes are important precursors to chiral building blocks (via asymmetric hydrogenation and other transformations)14 and intermediates in the synthesis of bioactive natural products.15 The stability of α-branched styrenes relative to the conjugated dienes obtained in our previous study enables the reactions to proceed with improved efficiency. Furthermore, the mild air- and moisture-tolerant conditions allow the reactions to tolerate aryl halide and allyl silane substrates that are often not compatible with traditional cross-coupling conditions (including the branched-selective Heck reactions noted above).
 | ||
| Scheme 2 Branched-selective aerobic oxidative Heck reactions with electronically unbiased alkenes. | ||
Our initial studies focused on assessing ligand effects on Pd(OAc)2-catalyzed coupling of 4-methoxyphenylboronic acid (1a) and 1-octene (2a) in tetrahydrofuran (THF). Reactions with monodentate pyridine ligands such as pyridine and 2-fluoropyridine exhibited a slight preference for linear product formation, albeit with low yields (<10% yield). Improved results were obtained with bidentate nitrogen ligands (Scheme 3). Higher yields were observed with 2,9-dimethyl-1,10-phenanthroline (dmphen) and 6,6′-dimethyl-2,2′-bipyridine which bear two methyl groups adjacent to the two nitrogen atoms. The branched product was favored in both of these reactions, with branched![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :linear selectivity up to ∼12:1 when the dmphen ligand was used. Related substituted-phen and -bpy ligands have been used previously in traditional and oxidative Heck-type coupling reactions; however, these precedents typically feature electronically biased substrates (e.g., vinyl amides or acrylates) in which the reactions exhibit product regioselectivity consistent with the substrate electronic effects.13
:linear selectivity up to ∼12:1 when the dmphen ligand was used. Related substituted-phen and -bpy ligands have been used previously in traditional and oxidative Heck-type coupling reactions; however, these precedents typically feature electronically biased substrates (e.g., vinyl amides or acrylates) in which the reactions exhibit product regioselectivity consistent with the substrate electronic effects.13
 | ||
| Scheme 3 Preliminary ligand screening showing the influence of ligands on regioselectivity. | ||
Cabri and co-workers have shown that Heck reactions that proceed via a “cationic pathway” exhibit enhanced selectivity for branched product formation with electron-rich alkenes owing to increased charge build-up in the alkene-insertion transition state.13b The cationic pathway can be favored over a less regioselective five-coordinate neutral pathway by using less basic counterions and/or more polar solvent to facilitate dissociation of the anionic ligand (Scheme 4).
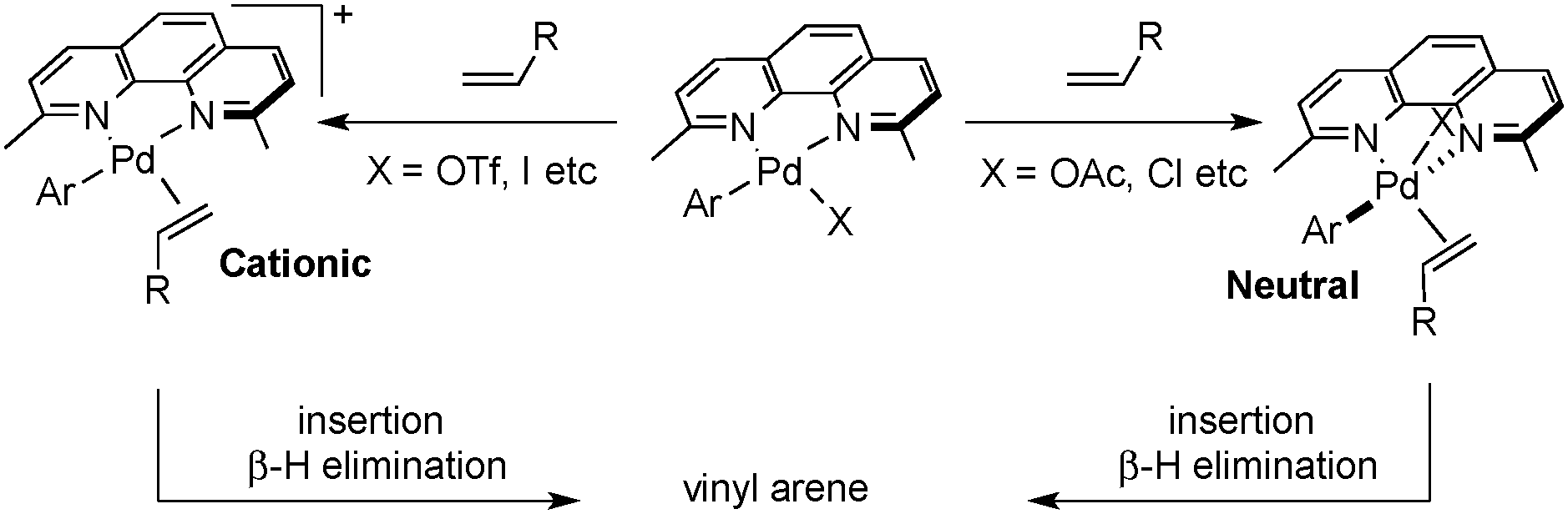 | ||
| Scheme 4 Cationic versus neutral, five-coordinate pathways. | ||
These considerations prompted us to test alternative counterions and solvents for the oxidative coupling of 1a and 2a. Selected results are summarized in Table 1. Up to 71% yield of the branched styrene product was obtained by replacing Pd(OAc)2 with Pd(TFA)2 and by changing the solvent from THF to N-methylpyrrolidone (NMP) (cf. entry 3, Table 1). The best results were obtained by using a 2:1 dmphen:Pd ratio, possibly reflecting catalyst decomposition, which results in lower reaction yields at a 1:1 ligand:Pd ratio (cf. entry 4). All reactions with Pd(TFA)2 as the PdII source exhibited high regioselectivity (>15:1), consistent with the involvement of the Cabri-like cationic pathway. The reactions performed in different solvents exhibited high regioselectivities in the presence of the Pd(TFA)2/dmphen catalyst system (entries 1–3). Assessment of acidic and basic additives (e.g., entries 5–7) did not improve the reaction outcome. Standard reaction conditions employed 1.5 equiv. of the boronic acid to account for the small amount of homocoupling side reaction; however, good product yields were also obtained by using the arylboronic acid as the limiting reagent with excess alkene (2 equiv.) (72% yield; cf. entries 3 and 9). Increasing the reaction temperature to 60 °C led to a slight improvement in the yield (entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Pd/dmphen (x/y mol %) | Additiveb | Solvent | Yieldc (%) |
|
a Reaction conditions: 1a (0.3 mmol), 2a (0.2 mmol), NMP (0.5 mL), 6 h.
b
y mol% of the additives used.
c GC yield based on 2a (or limiting reagent). Regioselectivity (branched:linear) >15:1.
d
1a/2a = 0.2 mmol/0.2 mmol.
e
1a/2a = 0.2 mmol/0.4 mmol.
f Reaction performed at 60 °C. NMM = N-methyl morpholine. NMP = N-methyl pyrrolidone.
|
||||
| 1 | 5/10 | — | Dioxane | 55 |
| 2 | 5/10 | — | CH3CN | 13 |
| 3 | 5/10 | — | NMP | 71 |
| 4 | 10/10 | — | NMP | 43 |
| 5 | 5/10 | KF | NMP | 57 |
| 6 | 5/10 | PhCO2H | NMP | 62 |
| 7 | 5/10 | NMM | NMP | 60 |
| 8d | 5/10 | — | NMP | 60 |
| 9e | 5/10 | — | NMP | 72 |
| 10e,f | 5/10 | — | NMP | 75 (70) |

The additive-free reaction conditions identified above were then used to explore the substrate scope. Before performing tests with a broad substrate scope, several substrate partners were tested at different ratios between the arylboronic acid and alkene (1.5:1, 1:1, and 0.5:1). In general, the results at 1.5:1 substrate ratio afforded the best product yields, owing to the formation of small amounts of homocoupled biaryls from the boronic acid substrate (see Table S1 in the ESI† for details). The reactivities of a variety of arylboronic acids were tested with diverse alkenes, and the products are displayed in Table 2 according to the different functional groups present in the alkene. Except where indicated otherwise, the reactions were performed with 1.5 equiv. of boronic acid relative to the alkene. All of the yields were obtained following isolation and purification, and two of the products (3c and 3i) were prepared on 1 mmol scale to confirm the reliability of the reactions.
|
|
|---|
|
a Reaction conditions: Pd(TFA)2 (0.01 mmol), dmphen (0.02 mmol), NMP (0.5 mL), 60 °C, 6 h. For 3b, 3e–3f, 3o: 1/2 = 0.2 mmol/0.4 mmol. For other substrates: 1/2 = 0.3 mmol/0.2 mmol.
b Isolated yield.
c Ratio of branched:linear was determined by 1H NMR spectroscopy analysis of crude reaction mixture.
d Isolated as a mixture of branched and linear (∼10%) regioisomers.
e Isomerization (∼10%) to the internal alkenes occurred during purification.
f Reaction in a round-bottom flask under O2 balloon.
|

|

The alkyl and phenyl containing alkenes reacted effectively with phenylboronic acids bearing p-MeO, -H, and -Cl substituents (3a–3d), with regioselectivity ranging from 6:1 (3c) to 15:1 (3a). Alkenes with ester moieties were even more effective, with yields typically above 80% (3e–3g). A free carboxylic acid was also well-tolerated in this reaction (3i). Three olefins with ether functionality, including tert-butyl vinyl ether, were demonstrated with electron deficient (p-bromophenyl) and electron-rich (2-furanyl, and 3,4,5-trimethoxyphenyl) boronic acids (3j–3l). It is worth noting that the bromide substituent derived from the arylboronic acid in 3j (similar to the aryl chloride in 3d) is tolerated owing to the mildness of the reaction conditions relative to typical Pd(0)-catalyzed reactions. The electron-rich tert-butyl vinyl ether substrate reacts exclusively with “branched” selectivity, as expected, and the resulting product then hydrolyzes in situ to give the aryl methyl ketone 3l.16 Two alkenes with free alcohol groups proceed in modest yields (3m and 3n). In the case of the tertiary alcohol 2-methyl-3-buten-2-ol, the product derives from linear-selective alkene functionalization, following by spontaneous dehydration to afford the conjugated diene 3n. The switch in regioselectivity probably arises primarily from a steric effect of the fully substituted carbon center adjacent to the alkene. In contrast, an allyl silane with the sterically hindered triisopropylsilyl substituent (TIPS) proceeds in good yield (72%, 3o) and excellent regioselectivity (20:1). In this case, the electron-donating beta-silicon effect, which should promote branched selectivity, presumably overrides any steric effect favoring the linear insertion product. This reactivity is quite appealing in light of the synthetic utility of substituted allyl silanes. Good results were also obtained with alkenes bearing remote siloxy groups (3p–3r), although the presence of the free phenol in the boronic acid used to afford 3p appears to attentuate the product yield. An electronically biased vinyl amide affords the branched product 3s in very good yield and regioselectivity, as expected, and remote phthalimide and secondary sulfonamide substituents were well tolerated (3t, 3u). Alkenes with pendant ketones (a methyl ketone and cyclohexanone) also were effective substrates (3v, 3w), and did not appear to be complicated by Pd-enolate formation (e.g., which could lead to dehydrydrogenation17). Styrene and butyl acrylate are electronically biased alkenes that typically favor linear product formation. The linear product remains strongly preferred with our catalyst system in the case of butyl acrylate (3y:95% yield with >20:1 regioselectivity), but styrene affords a 1:1 mixture of regioisomeric products (3x).
Factors that govern regioselectivity in Heck-type coupling reactions have been discussed extensively in the literature.2c,13b Bidentate ligands lead to cationic [PdII(L2)(aryl)(alkene)]+ intermediates that exhibit higher branched selectivity relative to neutral PdII(L)(X)(aryl)(alkene) intermediates formed with monodentate ligands. The cationic charge enhances charge build-up in the alkene-insertion transition state, thereby favoring the branched (Markovnikov) product. Access to the cationic intermediate noted in Scheme 4 is facilitated by use of the trifluoroactate anion, which may be readily displaced by an alkene.18 Electronic effects are not sufficient to explain the present results, however, because bidentate ligands lacking the methyl groups exhibit little selectivity for the branched product (cf.Scheme 3). Thus, the high regioselectivity is best rationalized by synergistic electronic and steric effects, whereby the methyl groups in the dmphen ligand sterically inhibit formation of the linear coupling product, as depicted in Scheme 5.
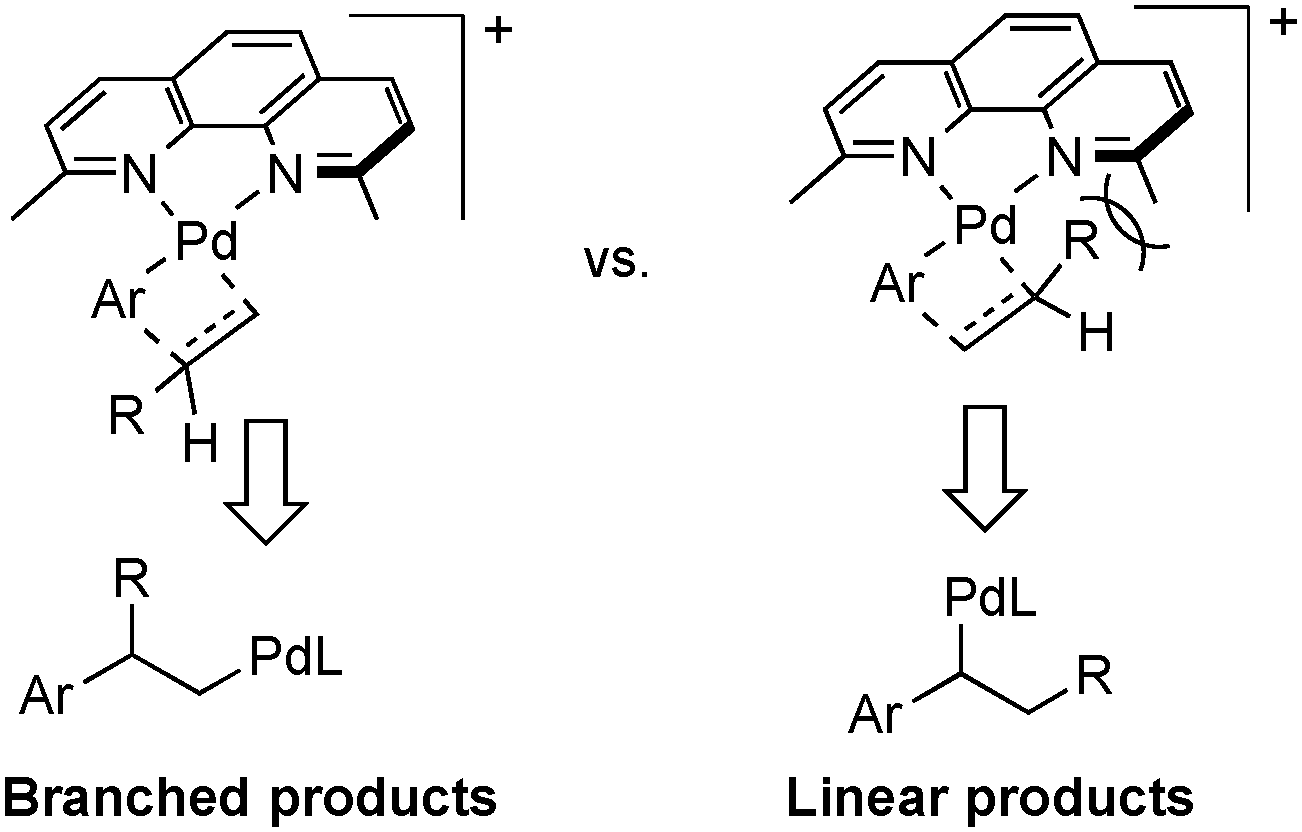 | ||
| Scheme 5 Transition state structures showing the steric contributions to branched vs. linear product formation. | ||
In this study, we have identified a readily accessible PdII catalyst system for aerobic oxidative coupling of arylboronic acids and electronically unbiased alkenes. The catalyst enables highly regioselective formation of α-substituted vinylarenes with substrates bearing diverse functional groups. The ease of operation and broad substrate scope make this method readily accessible and highly appealing for synthetic applications.
We are grateful for financial support from the NIH (R01 GM67173), Shanghai Institute of Organic Chemistry (SIOC Postdoctoral Fellowship for CWZ) for financial support of this work.
Notes and references
- For reviews, see: (a) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed; (b) M. Shibasaki, E. M. Vogl and T. Ohshima, Adv. Synth. Catal., 2004, 346, 1533 CrossRef CAS PubMed; (c) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed.
- For reviews, see: (a) R. F. Heck, Acc. Chem. Res., 1979, 12, 146 CrossRef CAS; (b) W. Cabri and I. Candiani, Acc. Chem. Res., 1995, 28, 2 CrossRef CAS; (c) P. Nilsson, K. Olofsson and M. Larhed, in The Mizoroki–Heck Reaction, ed. M. Oestreich, John Wiley & Sons, Chichester, 2009, pp. 133–162 Search PubMed; (d) B. Karimi, H. Behzadnia, D. Elhamifar, P. F. Akhavan, F. K. Esfahani and A. Zamani, Synthesis, 2010, 1399 CrossRef CAS PubMed.
- (a) Y. C. Jung, R. K. Mishra, C. H. Yoon and K. W. Jung, Org. Lett., 2003, 5, 2231 CrossRef CAS PubMed; (b) M. M. S. Andappan, P. Nilsson and M. Larhed, Chem. Commun., 2004, 218 RSC; (c) J. Lindh, P. Enquist, Å. Pilotti, P. Nilsson and M. Larhed, J. Org. Chem., 2007, 72, 7957 CrossRef CAS PubMed; (d) J. Ruan, X. Li, O. Saidi and J. Xiao, J. Am. Chem. Soc., 2008, 130, 2424 CrossRef CAS PubMed; (e) J. Ruan, J. A. Iggo, N. G. Berry and J. Xiao, J. Am. Chem. Soc., 2010, 132, 16689 CrossRef CAS PubMed.
- See ref. 3d and e and the following: (a) P. Harrison and G. Meek, Tetrahedron Lett., 2004, 45, 9277 CrossRef CAS PubMed; (b) M. M. S. Andappan, P. Nilsson, H. V. Schenck and M. Larhed, J. Org. Chem., 2004, 69, 5212 CrossRef CAS PubMed; (c) J. Mo, L. Xu and J. Xiao, J. Am. Chem. Soc., 2005, 127, 751 CrossRef CAS PubMed; (d) A. L. Hansen and T. Skrydstrup, J. Org. Chem., 2005, 70, 5997 CrossRef CAS PubMed; (e) J. Mo and J. Xiao, Angew. Chem., Int. Ed., 2006, 45, 4152 CrossRef CAS PubMed.
- H. A. Dieck and R. F. Heck, J. Am. Chem. Soc., 1974, 96, 1133 CrossRef CAS.
- (a) J. H. Delcamp and M. C. White, J. Am. Chem. Soc., 2006, 128, 15076 CrossRef CAS PubMed; (b) J. H. Delcamp, A. P. Brucks and M. C. White, J. Am. Chem. Soc., 2008, 130, 11270 CrossRef CAS PubMed; (c) E. W. Werner and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 13981 CrossRef CAS PubMed; (d) E. W. Werner and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 9692 CrossRef CAS PubMed.
- For examples of branched-selective Heck coupling with minimally biased substrates, such as allyl and homoallyl alcohols, see: (a) K. Olofsson, M. Larhed and A. Hallberg, J. Org. Chem., 1998, 63, 5076 CrossRef CAS; (b) J. Mo, L. Xu, J. Ruan, S. Liu and J. Xiao, Chem. Commun., 2006, 3591 RSC.
- (a) L. N. Qin, X. F. Ren, Y. P. Lu, Y. X. Li and J. R. Zhou, Angew. Chem., Int. Ed., 2012, 51, 5915 CrossRef CAS PubMed; (b) L. N. Qin, H. Hirao and J. R. Zhou, Chem. Commun., 2013, 49, 10236 RSC . For α-arylation of vinylarenes, see:; (c) Y. J. Zou, L. N. Qin, X. F. Ren, Y. P. Lu, Y. X. Li and J. R. Zhou, Chem. – Eur. J., 2013, 19, 3504 CrossRef CAS PubMed.
- S. Z. Tasker, A. C. Gutierrez and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 53, 1858 CrossRef CAS PubMed.
- For Ni-catalyzed branched-selective benzylation of alkenes, see: (a) R. Matsubara, A. C. Gutierrez and T. F. Jamison, J. Am. Chem. Soc., 2011, 133, 19020 CrossRef CAS PubMed; (b) E. A. Standley and T. F. Jamison, J. Am. Chem. Soc., 2013, 135, 1585 CrossRef CAS PubMed.
- (a) A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851 CrossRef CAS PubMed; (b) Y. Izawa, C. Zheng and S. S. Stahl, Angew. Chem., Int. Ed., 2013, 52, 3672 CrossRef CAS PubMed; (c) D. Wang, Y. Izawa and S. S. Stahl, J. Am. Chem. Soc., 2014, 136, 9914 CrossRef CAS PubMed.
- C. Zheng, D. Wang and S. S. Stahl, J. Am. Chem. Soc., 2012, 134, 16496 CrossRef CAS PubMed.
- For leading previous applications of dmphen as a ligand in oxidative and non-oxidative Heck reactions, see ref. 3b, c, 4a and the following: (a) W. Cabri, I. Candiani, A. Bedeschi and R. Santi, Synlett, 1992, 871 CrossRef CAS; (b) W. Cabri, I. Candiani and A. Bedeschi, J. Org. Chem., 1993, 58, 7421 CrossRef CAS; (c) P. A. Enquist, P. Nilsson, P. Sjoeberg and M. Larhed, J. Org. Chem., 2006, 71, 8779 CrossRef CAS PubMed; (d) P. A. Enquist, J. Lindh, P. Nilsson and M. Larhed, Green Chem., 2006, 8, 338 RSC; (e) K. S. Yoo, C. H. Yoon and K. W. Jung, J. Am. Chem. Soc., 2006, 128, 16384 CrossRef CAS PubMed; (f) J. O'Neill, K. S. Yoo and K. W. Jung, Tetrahedron Lett., 2008, 49, 7307 CrossRef PubMed; (g) A. Nordqvist, C. Bjoerkelid, M. Andaloussi, A. M. Jansson, S. L. Mowbray, A. Karlen and M. Larhed, J. Org. Chem., 2011, 76, 8986 CrossRef CAS PubMed.
- For selected examples, see: (a) R. Corberán, N. W. Mszar and A. H. Hoveyda, Angew. Chem., Int. Ed., 2011, 50, 7079 CrossRef PubMed; (b) R. Zhu and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 12655 CrossRef CAS PubMed; (c) X. Sun, L. Zhou, C.-J. Wang and X. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2623 CrossRef CAS PubMed; (d) J. Chen, B. Cheng, M. Cao and Z. Lu, Angew. Chem., Int. Ed., 2015, 54, 4661 CrossRef CAS PubMed.
- For selected examples, see: (a) S. P. Cook, A. Polara and S. J. Danishefsky, J. Am. Chem. Soc., 2006, 128, 16440 CrossRef CAS PubMed; (b) M. B. Johansen and M. A. Kerr, Org. Lett., 2008, 10, 3497 CrossRef CAS PubMed; (c) T. Ozawa, M. Kanematsu, H. Yokoe, M. Yoshida and K. Shishido, J. Org. Chem., 2012, 77, 9240 CrossRef CAS PubMed; (d) M. Wu and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 9759 CrossRef CAS PubMed.
- Similar reactivity has been observed previously, for examples, see ref. 3c and d, 4b and c and the following: (a) W. Cabri, I. Candiani, A. Bedeschi and R. Santi, J. Org. Chem., 1990, 55, 3654 CrossRef CAS; (b) T. M. Gøgsig, J. Kleimark, S. O. N. Lill, S. Korsager, A. T. Lindhardt, P.-O. Norrby and T. Skrydstrup, J. Am. Chem. Soc., 2012, 134, 443 CrossRef PubMed.
- Y. Izawa, D. Pun and S. S. Stahl, Science, 2011, 333, 209 CrossRef CAS PubMed.
- The ability of trifluoroacetate to promote formation of a cationic intermediate relative to acetate as an anionic ligand was the subject of considerable focus in the following study: A. B. Weinstein and S. S. Stahl, Angew. Chem., Int. Ed., 2012, 51, 11505 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data for the products and copies of NMR spectra. See DOI: 10.1039/c5cc05312a |
| This journal is © The Royal Society of Chemistry 2015 |