
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Resonance-assisted stabilisation of hydrogen bonds probed by NMR spectroscopy and path integral molecular dynamics†
M.
Dračínský
*a,
L.
Čechová
ab,
P.
Hodgkinson
c,
E.
Procházková
a and
Z.
Janeba
a
aInstitute of Organic Chemistry and Biochemistry, AS CR, Flemingovo nám. 2, 166 10 Prague, Czech Republic. E-mail: dracinsky@uochb.cas.cz
bDepartment of Chemistry of Natural Compounds, Institute of Chemical Technology Prague, Technická 5, 166 28 Prague, Czech Republic
cDepartment of Chemistry, Durham University, South Road, DH1 3LE, Durham, UK
First published on 23rd July 2015
Abstract
Path integral molecular dynamics and experimental NMR data are used to investigate resonance-assisted hydrogen bonds (RAHBs). When nuclear delocalisation is included in chemical shift calculations, the agreement with experiment is excellent, while static calculations show very poor performance. The results support the concept of RAHB, which has recently been questioned.
Hydrogen bonds play a key role in determining the shapes, properties and functions of biomolecules. Intermolecular hydrogen bonding is crucial for encoding genetic information (DNA base pairs) and interactions of biomolecules; intramolecular hydrogen bonding is pivotal to the formation of three-dimensional structures of biomacromolecules. The ability to form intramolecular hydrogen bonds (IMHBs) also has a great impact on the molecular structure and physico-chemical properties of small organic molecules, making the closed form (with an IMHB) more lipophilic than the open form.1 Furthermore, the cyclic system formed due to the presence of an IMHB can mimic the structural motifs of other biologically relevant molecules. Thus, replacing the real ring with a pseudoring with IMHB in drug-like molecules has become an important medicinal chemistry strategy.2
High stability of IMHBs has often been observed in planar systems where a six-membered ring is formed and the linker atoms are sp2-hybridized (as in amides, enols of β-diketones and β-enaminones, heteroaromatic rings, etc.).1 The high stability of these intramolecular hydrogen bonds has been termed resonance-assisted hydrogen bonding (RAHB), and was rationalized by enhanced π-delocalization involving the hydrogen atom (Fig. 1).3 Usually, RAHBs are classified as π-conjugated ring or chain motifs, for which characteristic changes in geometrical or electronic properties are observed, that is, elongation of formally double bonds and shortening of formally single bonds, together with the elongation of the X–H and shortening of the H⋯Y bond (X and Y stand for the H-bond donor and acceptor, respectively),4i.e. RAHBs are formally a linear combination of two tautomeric forms. RAHB has recently been reviewed.5 The concept of RAHB has, however, been criticized.6 For example, it was found in a series of enols of β-diketones and β-enaminones that the RAHB effect was not the primary reason behind the strength of their intramolecular hydrogen bonds, which was simply a consequence of the structure of the σ-skeleton of the system that keeps the hydrogen-bond donor and acceptor coplanar and closer to each other.6a However, regardless of the criticism, the concept of RAHB is still widely used in discussions of IMHB stability.
 | ||
| Fig. 1 The interrelation between the resonance-assisted hydrogen bond and π-electron delocalisation. | ||
Conjugated nitrosoamines can form planar systems with enhanced π-delocalization and, thus, belong to a class of compounds able to form strong IMHBs.7 During recent studies of 5-nitrosopyrimidines bearing distinct amino substituents in C4 and C6 positions, two possible rotamers, stabilized by strong IMHBs, were identified as two sets of signals in 1H NMR spectra.8 These 5-nitrosopyrimidines were proposed to work as purine mimics. The ratio of the two rotamers was found to be significantly substituent-dependent,8a with a broad range of conformation ratios, and the isomerisation barrier was found to be unexpectedly large (>22 kcal mol−1). It was even possible to separate the rotamers by column chromatography in some cases. This phenomenon has been termed planamerism.9
As RAHB involves the positioning of hydrogen atoms, the structure and properties of molecules with RAHB provided by static quantum-chemical calculations (geometry-optimised structures) may not be appropriate. Nuclear quantum effects (NQEs), such as zero-point vibrations and tunnelling, may be crucial in the case of hydrogen atoms. One route to including quantum effects in quantum-chemical simulations is provided by the formalisms based on Feynman's path integral10 (PI) approach. This path integral approach coupled with Car–Parrinello molecular dynamics has been employed in two purely theoretical studies of intramolecular hydrogen bonds in the solid state.11 Both studies indicated that the nuclear quantum effects led to a significant delocalisation of the bridging proton positions. One easily detectable manifestation of the quantum nature of nuclei is deuterium isotope effects in NMR spectra; zero-point fluctuations lead to differences in vibrationally averaged properties of compounds with the lighter and heavier hydrogen isotope. The small size of isotope effects, however, makes them challenging tests of ab initio calculations.12
Here we use a combination of path integral molecular dynamics (PIMD) with DFT to determine the nuclear quantum effects on the resonance-assisted stabilisation of intramolecular hydrogen bonds in two rotamers of 5-nitrosopyrimidine derivative 1 (Fig. 2), which differ in the orientation of the nitroso group. Both forms are stabilised by intramolecular hydrogen bonds and are present in solution in ca. 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio.8a We can expect that if resonance-stabilisation of hydrogen bonds exists, it will be more pronounced in the structure of rotamer 1A, where the pseudoring formed by IMHB forms part of a larger heteroaromatic conjugated system. Furthermore, a partial negative charge formed at the aniline nitrogen after a partial proton shift towards the NO group can be stabilised by delocalisation over two (hetero)aromatic rings. The calculations are compared with experimental NMR data measured for the equilibrium mixture of 15N-labelled compound 1. We demonstrate that nuclear delocalisation of hydrogen atoms has a significant impact on bond distances and 15N chemical shifts, with the effects being significantly more important in rotamer 1A. Furthermore, we observe unprecedentedly large through-hydrogen bond deuterium isotope effects on 15N chemical shifts of the nitroso group. The isotope effects could be rationalised in terms of changes in the molecular structure, and the PIMD approach enabled very good predictions of deuterium substitution effects on chemical shifts.
:3 ratio.8a We can expect that if resonance-stabilisation of hydrogen bonds exists, it will be more pronounced in the structure of rotamer 1A, where the pseudoring formed by IMHB forms part of a larger heteroaromatic conjugated system. Furthermore, a partial negative charge formed at the aniline nitrogen after a partial proton shift towards the NO group can be stabilised by delocalisation over two (hetero)aromatic rings. The calculations are compared with experimental NMR data measured for the equilibrium mixture of 15N-labelled compound 1. We demonstrate that nuclear delocalisation of hydrogen atoms has a significant impact on bond distances and 15N chemical shifts, with the effects being significantly more important in rotamer 1A. Furthermore, we observe unprecedentedly large through-hydrogen bond deuterium isotope effects on 15N chemical shifts of the nitroso group. The isotope effects could be rationalised in terms of changes in the molecular structure, and the PIMD approach enabled very good predictions of deuterium substitution effects on chemical shifts.
 | ||
| Fig. 2 The structure and atom numbering of two rotamers of compound 1. | ||
Proton-coupled and proton-decoupled nitrogen NMR spectra of compound 1 are shown in Fig. S1 in the ESI.† The equilibrium mixture of compound 1 contains ca. 7:3 ratio of rotamers 1A and 1B, which enables an easy assignment of NMR signals to the individual rotamers by comparing the signal intensities. A further advantage of working with the equilibrium mixture is that both rotamers of compound 1 are in one solution, avoiding errors caused by sample preparation or referencing of chemical shifts.
In further discussion, we concentrate on nitrogen chemical shifts of the nitroso group in both rotamers because the chemical shifts of the other atoms involved in the IMHB will be significantly affected by solvation, which is difficult to involve in quantum-chemical calculations of NMR parameters.13 The effect of solvation for most of the atoms in compound 1 is probably quite large, because the molecule has several acidic hydrogens and the experiments were done in DMSO, which is a good hydrogen bond acceptor. Unfortunately, the compound is poorly soluble in non-polar solvents; we were only able to acquire 1H NMR spectrum in CDCl3 on an 850 MHz spectrometer, which confirmed large solvent-induced shifts (Fig. S2 in the ESI†). On the other hand, the NO group cannot form hydrogen bonds with DMSO and we may expect that its solvation shell structure will be similar in both rotamer forms.
When the structure of compound 1 is optimised at the widely used B3LYP DFT level and shielding constants are calculated for the optimised structure, the calculated chemical shift difference of the nitroso nitrogen between rotamer 1A and 1B is far from the experimental value (Table 1). Similar discrepancy with experiment is also observed for other functionals (Table S1 in the ESI†).
| Static calculation (σ) | PIMD corrections | Experiment (δ) | |||||
|---|---|---|---|---|---|---|---|
| 6N–H | 4N–H | C6–N–H | C4–N–H | Total | |||
| 1A | −507.1 | +14.6 | +1.8 | +2.1 | +1.8 | −486.9 | 624.8 |
| 1B | −517.1 | +3.3 | +5.8 | −0.1 | +1.6 | −506.5 | 645.4 |
| Δδ(B–A) | 10.0 | 19.6 | 20.6 | ||||
Recently, we introduced an approach for including nuclear quantum effects in NMR calculations based on convoluting calculated shielding or coupling surfaces with probability distributions of selected bond distances and valence angles obtained from PIMD simulations.14Fig. 3 shows the probability distributions of the N–H bond distances observed during PIMD simulations. Probability distributions of other selected bond distances are shown in Fig. S3 in the ESI.† As expected, the hydrogen atoms are heavily delocalised in the PIMD simulations, leading to broad distance distributions. The PIMD distributions are also shifted towards longer bond lengths than those found in geometry-optimised structures, particularly in the case of rotamer 1A. Note that a simple one-dimensional energy scan (i.e. not including nuclear delocalisation) inverted to a Boltzmann distribution gives a much narrower probability distribution (Fig. S4, ESI†). We used the N–H bond distance distributions together with C–N–H angle distributions found in PIMD simulations of both rotamers for the calculations of PIMD-induced corrections of chemical shifts of the nitroso group. We calculated the dependence of the isotropic shieldings of compound 1 on the N–H bond distance and the C–N–H valence angle (Fig. S5, ESI†) and used the bond distance and valence angle probabilities to determine weighted averages of the 15N isotropic shieldings. The inclusion of the PIMD corrections leads to a very good agreement between experimental and calculated chemical shift differences in the two rotamers (Table 1). When the same procedure was used for probability distributions obtained from classical-nucleus MD (conventional MD), the calculated chemical shift difference was still more than 5 ppm off the experimental value (Table S2 in the ESI†).
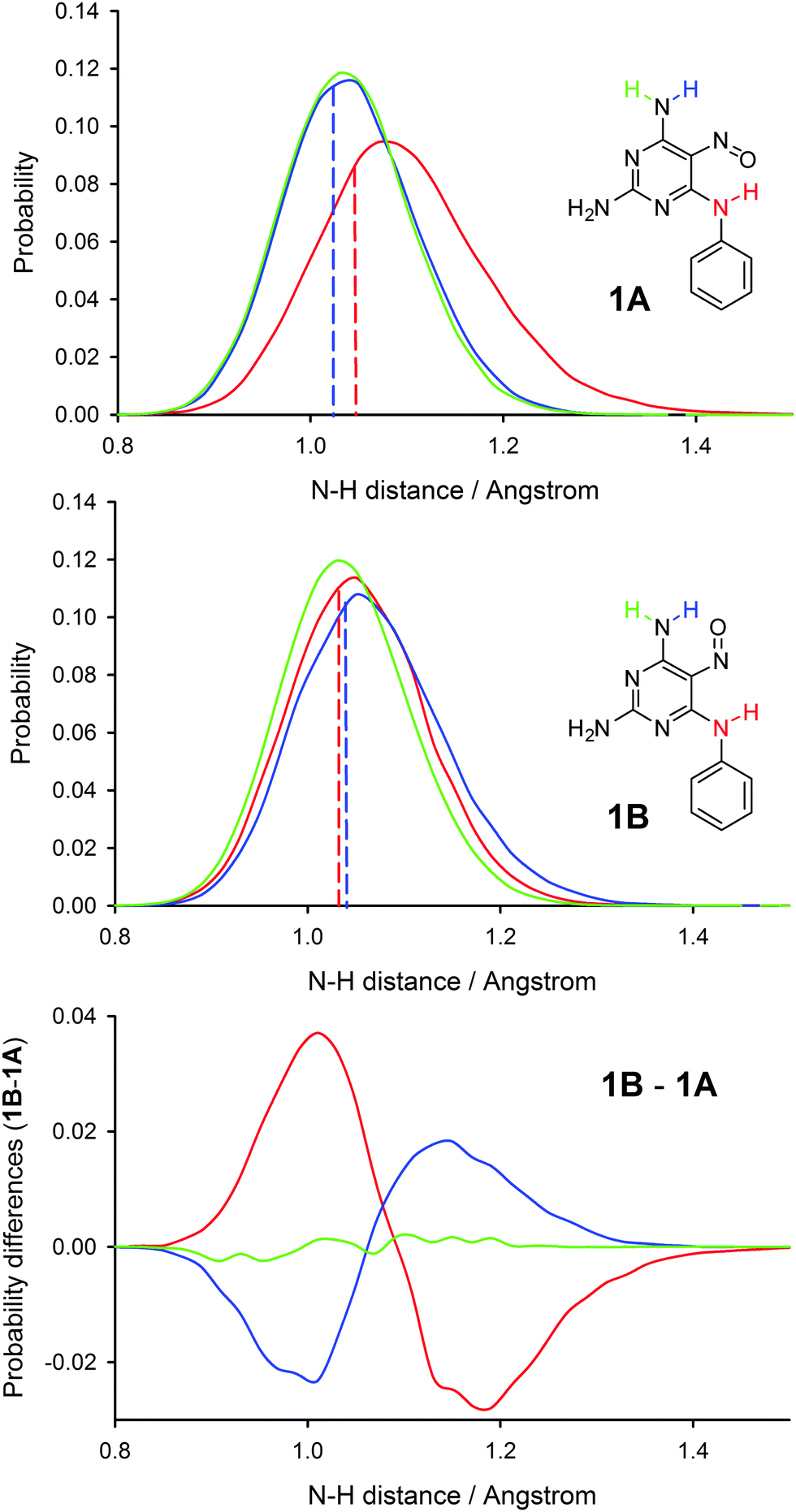 | ||
| Fig. 3 Probability distributions (sampled by 0.02 Å) and their differences in N–H bond distances obtained from PIMD simulations of both rotamers of compound 1. The dashed lines indicate the geometry optimised N–H distance. No rotation of the NH2 group around the C–N bond was observed during the PIMD simulations. Every 10 ps PIMD simulation took ca. 10 days on 128 computational cores. | ||
When comparing rotamer 1A and 1B, the following differences were found in the probability distributions of bond distances. The N–H distance in the amino group involved in the IMHB is longer and the distribution is broader in rotamer 1A, the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O distance is slightly longer in rotamer 1A, the C5–NO distance is slightly shorter in rotamer 1A, and the hydrogen bond distance O⋯H is significantly shorter in rotamer 1A. The average bond distances found in PIMD trajectories are shown in Table S3 in the ESI.† All these changes may be explained by a larger resonance stabilisation of the IMHB in rotamer 1A. The N–H bond distance differences upon rotamer exchange are shown in Fig. 3, which visualises clearly that the IMHB formation shifts the N–H bond distances towards larger values, with the effect being significantly larger in the case of the 6N–H bond involved in IMHB in rotamer 1A.
O distance is slightly longer in rotamer 1A, the C5–NO distance is slightly shorter in rotamer 1A, and the hydrogen bond distance O⋯H is significantly shorter in rotamer 1A. The average bond distances found in PIMD trajectories are shown in Table S3 in the ESI.† All these changes may be explained by a larger resonance stabilisation of the IMHB in rotamer 1A. The N–H bond distance differences upon rotamer exchange are shown in Fig. 3, which visualises clearly that the IMHB formation shifts the N–H bond distances towards larger values, with the effect being significantly larger in the case of the 6N–H bond involved in IMHB in rotamer 1A.
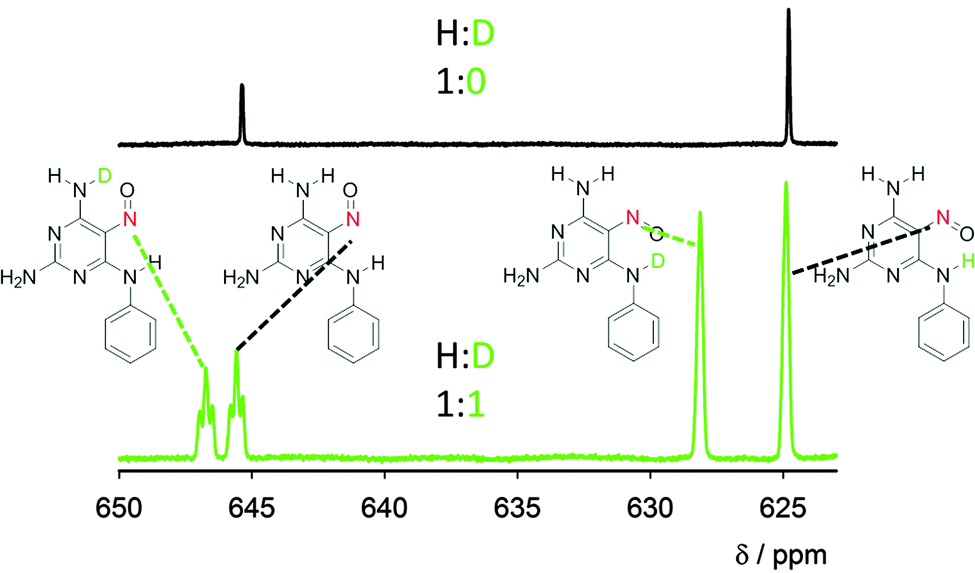 | ||
| Fig. 4 Nitroso group region of the 15N NMR spectrum of compound 1 in DMSO (black spectrum) and of compound 1 recrystallized from the H2O/D2O mixture (1:1) and dissolved in DMSO (green spectrum). The assignment of the signals is based on a comparison between the spectra with a different H:D ratio and spectra of similar model compounds (details in the ESI†). Expansion of the spectra with the signal assignments is depicted in the ESI† (Fig. S6). | ||
Experimental deuterium isotope effects on 15N chemical shifts were obtained for compound 1 recrystallised from a 1:1 mixture of H2O/D2O (Fig. 4). In rotamer 1A, an extraordinarily large (3.3 ppm) through-hydrogen-bond isotope effect is observable; normally, the isotope effect decreases rapidly with the number of bonds between the exchanged hydrogen and the observed nucleus, with one-bond 15N chemical shift changes (exchange of NH for ND) being 1.2–1.4 ppm.15 On the other hand, hydrogen to deuterium exchange in the free amino group (not involved in the IMHB) in position C4 of rotamer 1A leads to a minor line broadening of the nitroso nitrogen resonance, i.e. the isotope effect through four covalent bonds is negligible (Fig. 4, see Fig. S6, ESI,† for expansion). In rotamer 1B, the through-hydrogen-bond effect is significantly smaller (1.1 ppm) and through-covalent-bond effects in the order of 0.2 ppm are also observable. The unusually large isotope effects confirm large anharmonicity of the N–H bond potential in the IMHBs of both rotamers. However, the three times larger isotope shift in form 1A indicates larger geometry differences in this form upon isotope substitution, which can be explained by larger delocalisation of the lighter 1H atom than deuterium in rotamer 1A.
We expect that the effects of solvation on chemical shifts will be very similar for both the deuterated and non-deuterated compound; therefore, deuterium isotope shifts may be safely evaluated for other atoms in the molecule. The isotope-exchange effect on the 6-NH nitrogen in rotamer 1A is almost twice as large as in rotamer 1B (−1.26 ppm vs. −0.69 ppm, see Fig. S7 in the ESI† for experimental spectra), which confirms again large anharmonicity of the N–H potential when the hydrogen atom is involved in the IMHB. On the other hand, the isotope-exchange effect on 4-NH nitrogen is comparable in both forms 1A and 1B (−0.6 ppm vs. −0.7 ppm, exchange of one of the NH2 hydrogens only).
The PIMD simulations performed on both rotamers of deuterated compound 1 confirm the experimental data. From the geometrical changes upon deuteration (Table S3, ESI†) it follows that hydrogen to deuterium exchange leads to lower resonance stabilisation of the IMHB. Upon deuteration, the same trends in changes of intramolecular distances and angles are observed as when going from rotamer 1A to 1B, although the magnitude of the changes is lower, leading to ca. six times larger effect of the rotamer exchange than isotope exchange on the nitroso nitrogen shielding. Using just N–H distances and C–N–H valence angles to estimate deuterium isotope effects based on changes in probability distributions during the PIMD simulations, the predicted values are 4.9 and 1.6 ppm in the form 1A and 1B, respectively, which compares well with the experimental values (3.3 and 1.1 ppm).
In summary, we have proven by experimental 15N NMR data and PIMD simulations that resonance stabilisation of IMHBs is tightly related to nuclear quantum effects, namely delocalisation of hydrogen atoms involved in these IMHBs. Nuclear delocalisation is larger in conformer 1A, where all bond distances in the pseudoring formed by the IMHB also confirm larger resonance stabilisation than in conformer 1B. In deuterated compound 1, the delocalisation of the deuterium nucleus is smaller leading to lower resonance stabilisation of the IMHB (manifested by shortening of formally double bonds and elongation of formally single bonds) and large isotope shifts in the 15N NMR spectrum. Nuclear delocalisation has to be taken into account when describing the bonding in systems with strong hydrogen bonds, and is more important in structures where larger RAHB effects are anticipated. Conventional calculations (both geometry optimisation and classical-nucleus MD without NQEs) do not clearly show the changes in bond lengths that would be expected for RAHB and fail to reproduce chemical shifts in these systems. The better agreement of NMR data when nuclear delocalisation is included shows that RAHB effects are observable via NMR and supports the concept of resonance stabilisation of hydrogen bonds. The approach combining PIMD simulations with experimental NMR data is generally applicable for studies of both intra- and intermolecular hydrogen bonds.
The work has been supported by the Czech Science Foundation (grant no. 15-11223S).
Notes and references
- B. Kuhn, P. Mohr and M. Stahl, J. Med. Chem., 2010, 53, 2601 CrossRef CAS PubMed.
- (a) P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer, Bioorg. Med. Chem. Lett., 2008, 18, 897 CrossRef CAS PubMed; (b) C. N. Hodge and J. Pierce, Bioorg. Med. Chem. Lett., 1993, 3, 1605 CrossRef CAS; (c) P. Furet, G. Bold, F. Hofmann, P. Manley, T. Meyer and K. H. Altmann, Bioorg. Med. Chem. Lett., 2003, 13, 2967 CrossRef CAS; (d) K. A. Menear, C. Adcock, F. C. Alonso, K. Blackburn, L. Copsey, J. Drzewiecki, A. Fundo, A. Le Gall, S. Gomez, H. Javaid, C. F. Lence, N. M. B. Martin, C. Mydlowski and G. C. M. Smith, Bioorg. Med. Chem. Lett., 2008, 18, 3942 CrossRef CAS PubMed; (e) A. M. Lord, M. F. Mahon, M. D. Lloyd and M. D. Threadgill, J. Med. Chem., 2009, 52, 868 CrossRef CAS PubMed; (f) B. Osmialowski, E. Kolehmainen and M. Kowalska, J. Org. Chem., 2012, 77, 1653 CrossRef CAS PubMed.
- G. Gilli, F. Bellucci, V. Ferretti and V. Bertolasi, J. Am. Chem. Soc., 1989, 111, 1023 CrossRef CAS.
- (a) S. J. Grabowski, J. Phys. Org. Chem., 2004, 17, 18 CrossRef CAS PubMed; (b) M. Palusiak, S. Simon and M. Sola, J. Org. Chem., 2006, 71, 5241 CrossRef CAS PubMed.
- (a) S. Bolvig and P. E. Hansen, Curr. Org. Chem., 2000, 4, 19 CrossRef CAS; (b) L. Sobczyk, S. J. Grabowski and T. M. Krygowski, Chem. Rev., 2005, 105, 3513 CrossRef CAS PubMed; (c) K. A. Lyssenko and M. V. Antipin, Russ. Chem. Bull., 2006, 55, 1 CrossRef CAS.
- (a) P. Sanz, O. Mó, M. Yáñez and J. Elguero, J. Phys. Chem. A, 2007, 111, 3585 CrossRef CAS PubMed; (b) P. Sanz, O. Mó, M. Yáñez and J. Elguero, Chem.–Eur. J., 2008, 14, 4225 CrossRef CAS PubMed; (c) P. Sanz, O. Mó, M. Yáñez and J. Elguero, Phys. Chem. Chem. Phys., 2009, 11, 762 RSC; (d) F. Fuster and S. J. Grabowski, J. Phys. Chem. A, 2011, 115, 10078 CrossRef CAS PubMed; (e) R. Kurczab, M. P. Mitoraj, A. Michalak and T. Ziegler, J. Phys. Chem. A, 2010, 114, 8581 CrossRef CAS PubMed; (f) A. R. Nekoei and M. Vatanparast, New J. Chem., 2014, 38, 5886 RSC.
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405 CrossRef CAS.
- (a) E. Procházková, L. Čechová, Z. Janeba and M. Dračínský, J. Org. Chem., 2013, 78, 10121 CrossRef PubMed; (b) I. Susvilo, A. Brukstus and S. Tumkevicius, Tetrahedron Lett., 2005, 46, 1841 CrossRef CAS PubMed; (c) A. Marchal, M. Nogueras, A. Sanchez, J. N. Low, L. Naesens, E. De Clercq and M. Melguizo, Eur. J. Org. Chem., 2010, 3823 CrossRef CAS PubMed.
- L. Čechová, E. Procházková, I. Císařová, M. Dračínský and Z. Janeba, Chem. Commun., 2014, 50, 14892 RSC.
- R. P. Feynman and A. R. Hibbs, Quantum Mechanics and Path Integrals, McGraw-Hill, New York, 1965 Search PubMed.
- (a) P. Durlak and Z. Latajka, Phys. Chem. Chem. Phys., 2014, 16, 23026 RSC; (b) A. Jezierska and J. J. Panek, J. Comput. Chem., 2009, 30, 1241 CrossRef CAS PubMed.
- C. J. Jameson, in Encyclopedia of NMR, ed. R. K. Harris, John Wiley & Sons, Ltd, Chichester, 2012 Search PubMed.
- M. Dračínský and P. Bouř, J. Chem. Theory Comput., 2010, 6, 288 CrossRef.
- M. Dračínský and P. Hodgkinson, Chem.–Eur. J., 2014, 20, 2201 CrossRef PubMed.
- P. E. Hansen, Molecules, 2015, 20, 2405 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures and tables, synthesis and characterisation of the prepared compounds, computational and experimental methods. See DOI: 10.1039/c5cc05199a |
| This journal is © The Royal Society of Chemistry 2015 |