
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric cyclopropanation of conjugated cyanosulfones using a novel cupreine organocatalyst: rapid access to δ3-amino acids†
Lewis S. Aitken,
Lisa E. Hammond,
Rajkumar Sundaram,
Kenneth Shankland,
Geoffrey D. Brown and
Alexander J. A. Cobb*
School of Chemistry, Food and Pharmacy (SCFP), University of Reading, Whiteknights, Reading, Berks RG6 6AD, UK. E-mail: a.j.a.cobb@reading.ac.uk
First published on 21st July 2015
Abstract
An organocatalytic asymmetric synthesis of a novel, highly functionalised cyclopropane system furnished with versatile substituents and containing a quaternary centre is described. The process utilises a new bifunctional catalyst based on the cinchona alkaloid framework and the products made using this catalyst were obtained as single diastereoisomers, with very high enantioselectivities (up to 96% ee). We have also demonstrated that these resulting cyclopropanes are very useful synthetic intermediates to interesting products, such as the difficult to access δ3-amino acids.
The cyclopropane moiety is an important and common motif in a number of biologically active compounds and natural products.1 It is also of synthetic use, especially if functionalised with the correct arrangement of electron-donating and electron-withdrawing groups, as it can then undergo a variety of nucleophilic, electrophilic or pericyclic reactions.2 If the cyclopropane is asymmetric in nature, then the products of these transformations also tend to be enantiopure. As a result, routes to enantiopure cyclopropanes have been extensively studied,3 particularly with respect to the Simmons–Smith reaction,4 and transition metal catalysed reactions using carbene intermediates.5 Organocatalysis has also demonstrated its utility in their synthesis. Aggarwal,6 Gaunt,7and MacMillan8 have all utilized ylides to achieve this, whilst the Michael-initiated ring closure (MIRC) has also proven to be a powerful method of choice.9 In spite of this, MIRCs are dominated by compounds containing the nitro-group functionality, thus limiting substrate scope.10 Therefore, in order to increase the diversity of this class of reaction, we sought to use the versatile conjugated cyanosulfone 1 shown in Scheme 1 instead. Previously our group has demonstrated this substrate to be a useful Michael acceptor in an organocatalytic domino process,11 and so we wished to examine its utility in the MIRC cyclopropanation with bromomethylmalonate 2. We felt that the resulting novel cyclopropane product 3, which is highly functionalised and contains a quaternary stereogenic centre, could undergo a variety of transformations to give useful products.
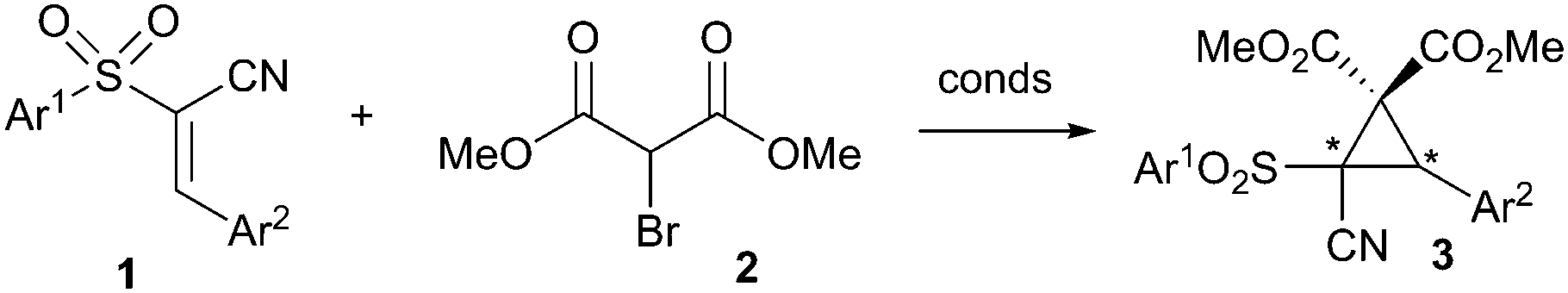 | ||
| Scheme 1 Concept – organocatalytic synthesis of a versatile cyclopropane. | ||
With this in mind, a test reaction was devised that utilised a variety of solvents and bifunctional catalysts (Table 1 for summary, ESI† for full investigation), mostly based around the framework of the cinchona alkaloids, which are well-known for their ability to activate electrophiles and pronucleophiles simultaneously.12 Unfortunately, the most well-established bifunctional organocatalyst type – the thiourea-derived systems – failed to catalyse the reaction under our screening conditions. However, the quinine-derived organocatalyst cupreine I was able to impart some enantioselectivity, though both this and the yield were low (entry 1). Encouragingly however, total diastereocontrol was observed in all cases. In an attempt to improve both enantioselectivity and yield, it was hypothesised that adding greater bulk to the secondary alcohol of the catalyst might improve the selectivity of the reaction. A number of cupreine-derived organocatalysts were therefore synthesised and screened (selected examples given, see ESI† for full study). In spite of the low yields obtained once again from this screen, we were encouraged to find that the acylated cupreine systems did indeed improve the stereoselectivity markedly (interestingly, the hydrocupreine derivatives performed better in terms of enantioselectivity than the cupreine derivatives), and that novel catalyst system VII performed best in this respect (entry 11).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat | Solvent | Yield, % | drb | Erc |
| a Reaction conditions: room temperature, 120 h. b Determined by 1H-NMR. c Determined by chiral HPLC using a Chiralpak AD-H stationary phase. | |||||
| 1 | I | CH2Cl2 | 10 | >19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
68:32 |
| 2 | I | THF | 18 | >19:1 |
59:41 |
| 3 | I | CHCl3 | 7 | >19:1 |
62:38 |
| 4 | I | PhMe | Trace | nd | nd |
| 5 | I | iPrOH | Trace | nd | nd |
| 6 | II | CH2Cl2 | 12 | >19:1 |
50:50 |
| 7 | III | CH2Cl2 | 21 | >19:1 |
54.5:45.5 |
| 8 | IV | CH2Cl2 | 17 | >19:1 |
79.5:20.5 |
| 9 | V | CH2Cl2 | 17 | >19:1 |
85:15 |
| 10 | VI | CH2Cl2 | 11 | >19:1 |
83.5:16.5 |
| 11 | VII | CH2Cl2 | 12 | >19:1 |
86:14 |
| 12 | VIII | CH2Cl2 | 31 | >19:1 |
50:50 |
| 13 | IX | CH2Cl2 | 8 | >19:1 |
50:50 |
| 14 | X | CH2Cl2 | 12 | >19:1 |
50:50 |
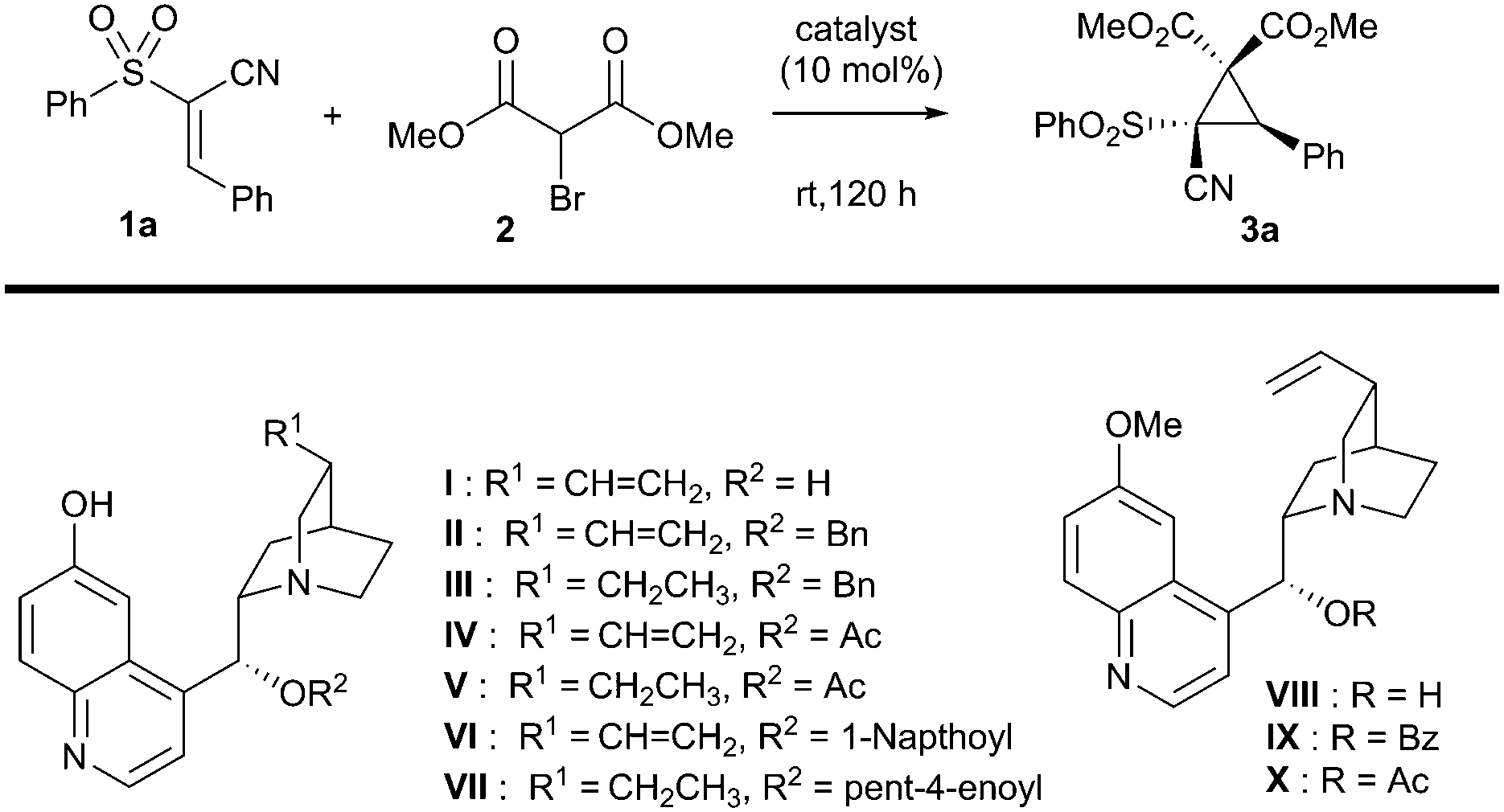
As with other studies using cupreine, it appears that the quinoline alcohol is critical to the enantioselectivity of the reaction, as if this is methylated, no selectivity occurs – although there is still comparable turnover (entries 12–14).13 Clearly, however, the yield of the reaction required improvement, and so we endeavoured to achieve this by addition of base which is purported to enhance the reaction by neutralising the liberated HBr.14 Owing to its marginally better selectivity profile, we screened a number of bases against organocatalyst VII, and pleasingly found that the yield improved significantly (Table 2), as did the rate of reaction. Impressively, we also found that the enantioselectivity was generally much better and that oven-dried potassium carbonate gave the best results in terms of this. These dry conditions are in contrast with a related study where water was shown to enhance the reaction.14 Our final optimisation looked at the effect of catalyst loading and temperature, where ultimately, a loading of 10 mol% at the reduced temperature of −10 °C for 96 h in dichloromethane was deemed to be the best conditions for this cyclopropanation reaction (entry 7 – see ESI† for full study).
|
|
||||
|---|---|---|---|---|
| Entry | Baseb | Eq | Yield, % | Er |
| a In all cases only one diastereoisomer was observed by 1H NMR. b All bases presented in this table were dried in a vacuum oven before use. c Reaction performed at −10 °C for 96 h. | ||||
| 1 | Na2CO3 | 1 | 92 | 85:15 |
| 2 | Cs2CO3 | 1 | 86 | 53.5:46.5 |
| 3 | K2CO3 | 0.5 | 88 | 86:14 |
| 4 | K2CO3 | 1 | 90 | 85:15 |
| 5 | K2CO3 | 2 | 92 | 84.5:15.5 |
| 6 | K2CO3 | 4 | 90 | 83.5:16.5 |
| 7c | K2CO3 | 1 | 92 | 88:12 |
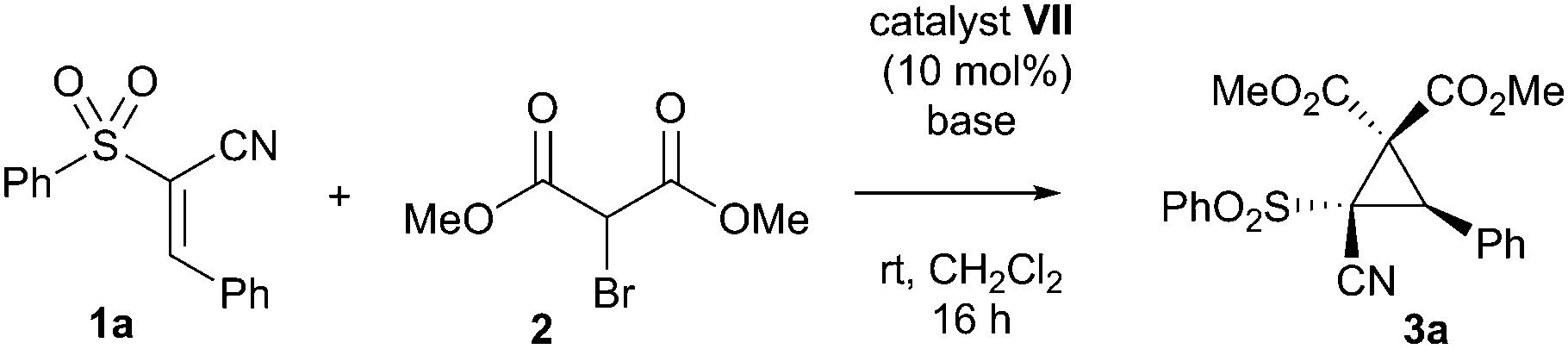
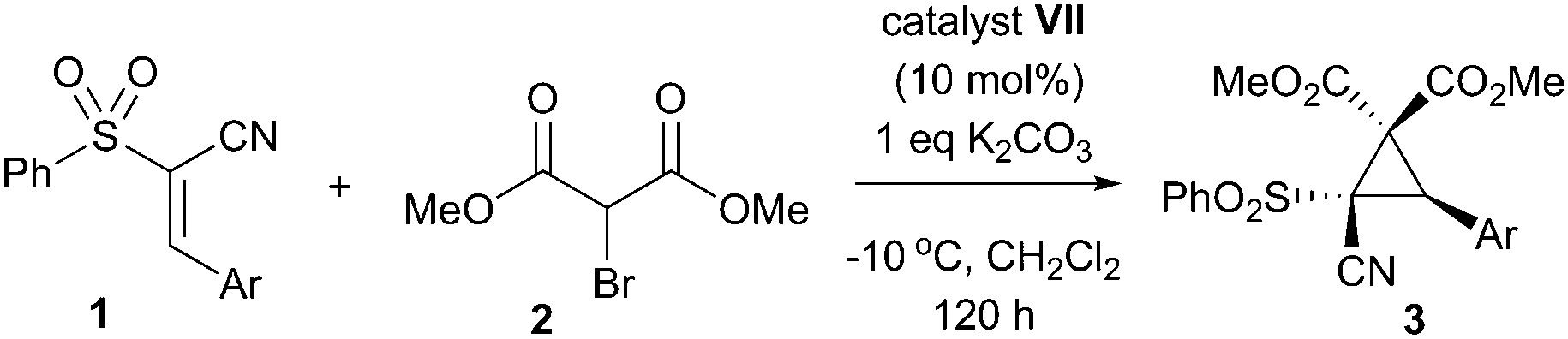
With an effective protocol for the enantioselective cyclopropanation in hand, we examined the generality and scope of the methodology. A variety of conjugated cyanosulfones were studied which varied in the electronics of the aryl group (Table 3). In most cases the initial enantioselectivity was good. More pleasingly, however was the fact that the majority of these could be significantly improved through a single recrystallization. In all cases the diastereoselectivity was excellent and the yields were also excellent. The absolute crystal structure of 3c was determined by X-ray crystallography (Fig. 1) and the other products assigned by analogy.
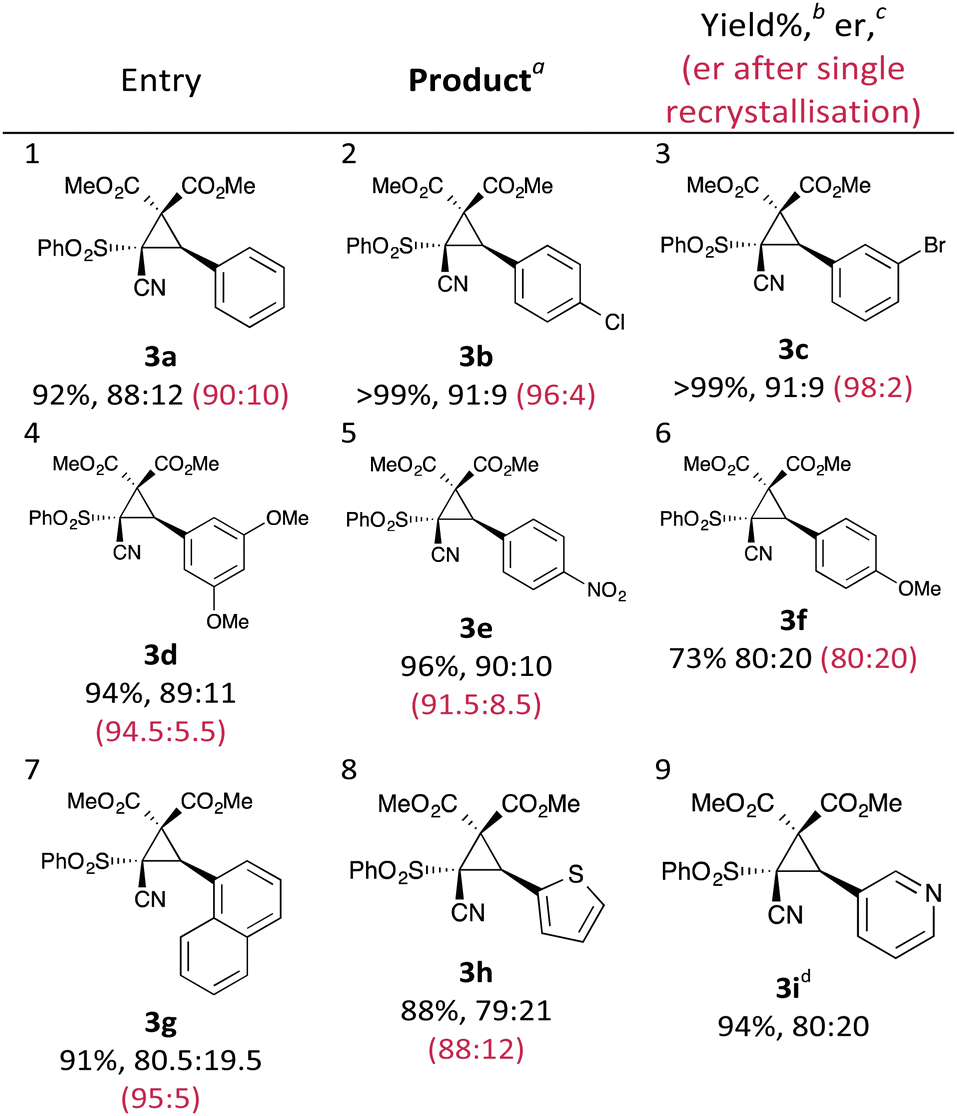
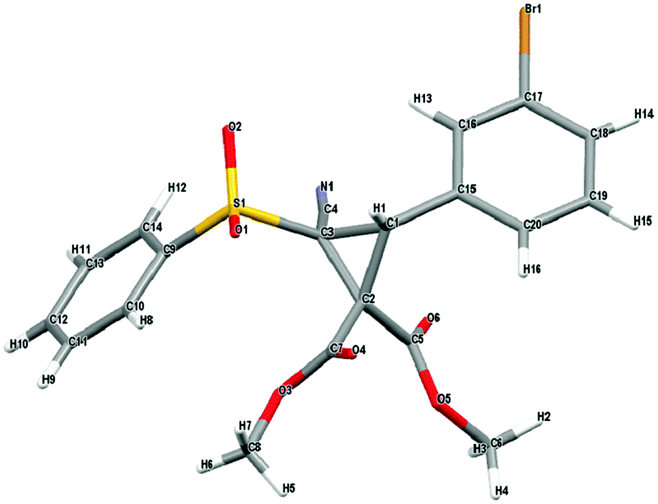 | ||
| Fig. 1 X-ray crystal structure of 3c, showing the absolute configuration. CCDC 1403683. | ||
We speculate that as with all bifunctional organocatalytic systems, the reaction proceeds via simultaneous activation of the pronucleophile (bromomalonate) and the electrophilic cyanosulfone through co-ordination to the cupreine derived catalyst. We tentatively propose that the transition state shown in Fig. 2 could account for this activation mode and the resulting stereochemistry. We also believe that the exceptionally high diastereoselectivity is due to the intermediate anion probably being long-lived enough to equilibrate to the thermodynamically more stable conformation, whereby the sulfone and the aryl group adopt positions that avoid unfavourable gauche interaction.
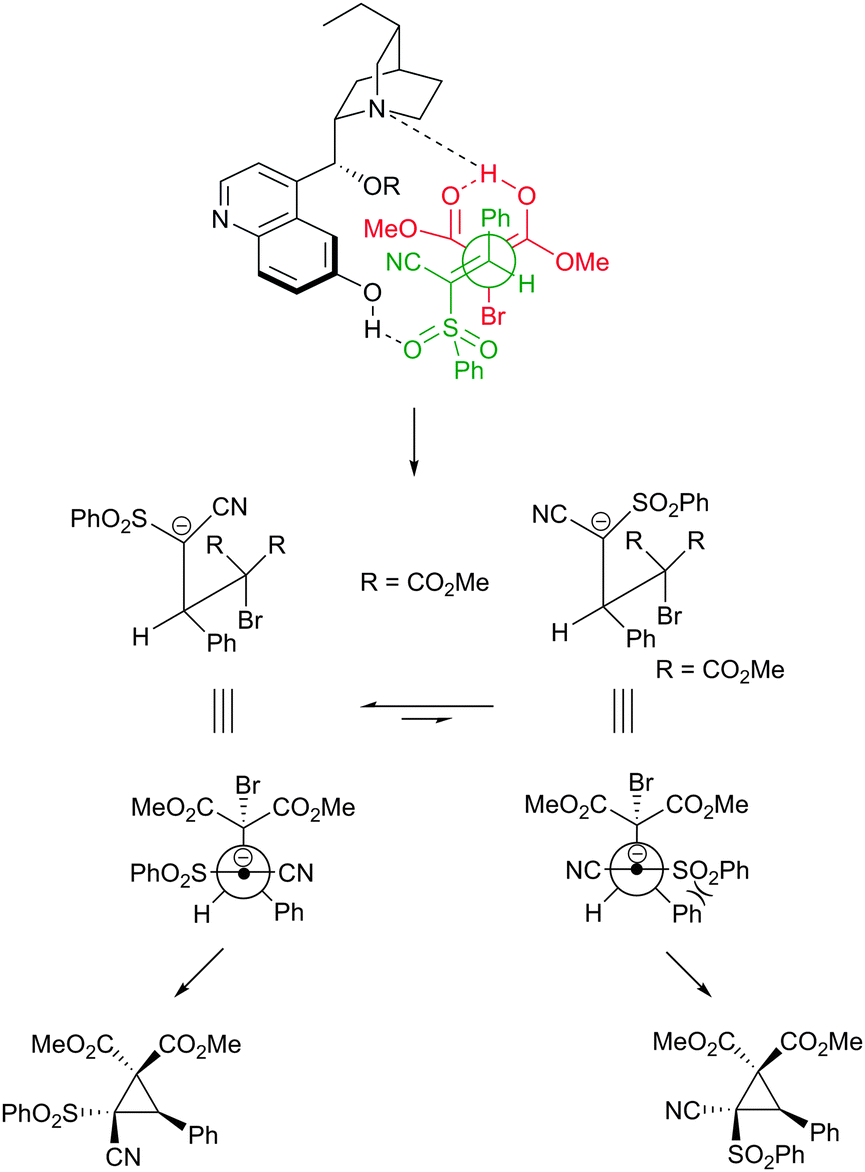 | ||
| Fig. 2 Proposed transition state and rationale for observed diastereoselectivity. | ||
Finally, in order to demonstrate the utility of these compounds, we subjected them to a variety of transformative conditions. To begin with, we attempted a straightforward desulfonylation using refluxing magnesium in methanol15 to obtain the δ3-amino acid precursor 6 with no depreciation of enantiomeric ratio. We propose that this occurs via a radical desulfonylation, generating cyclopropane radical 4 which rapidly ring opens to the radical anion 5. Protonation, followed by addition of a further electron to the malonate radical and then a second protonation gives the δ3-amino acid precursor 6.16 Hydrolysis/decarboxylation and reduction of the nitrile then gives the corresponding δ3-amino acid (Scheme 2).
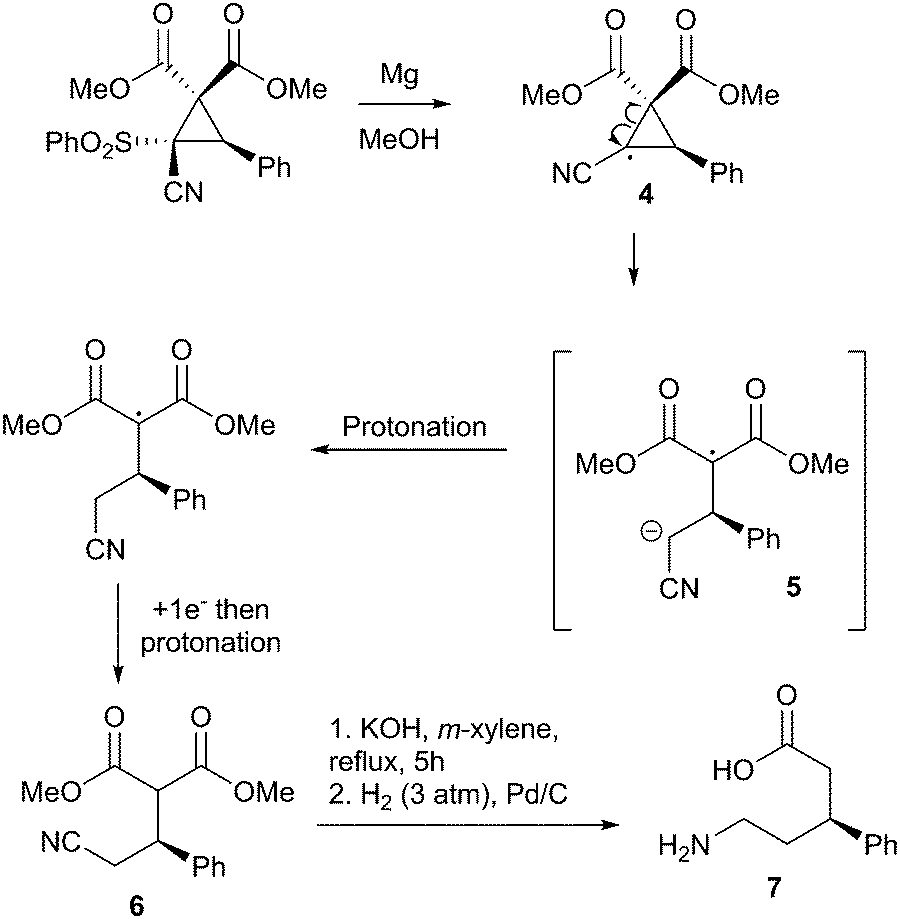 | ||
| Scheme 2 Proposed magnesium-initiated ring-opening to form a δ3-amino acid precursor. | ||
This is an intriguing result, as reports on the synthesis of enantiopure δ-amino acids appear to be extremely rare in the literature. Our methodology represents a rapid new way of accessing this particular class, being just five linear steps.17
Finally, we also achieved the synthesis of the intriguing 3-azabicyclo[3.1.0]hexane system 8 through the borane mediated reduction of the nitrile, leading to concomitant cyclisation of the resulting amine onto the syn-ester followed by in situ reduction of the γ-lactam to the fused pyrrolidine system (Scheme 3). This fascinating ring system appears in a wide variety of natural products and medicinally useful compounds, such as the ergot alkaloid, cycloclavine 9 and the bioengineered antibiotic indolizomycin 10.
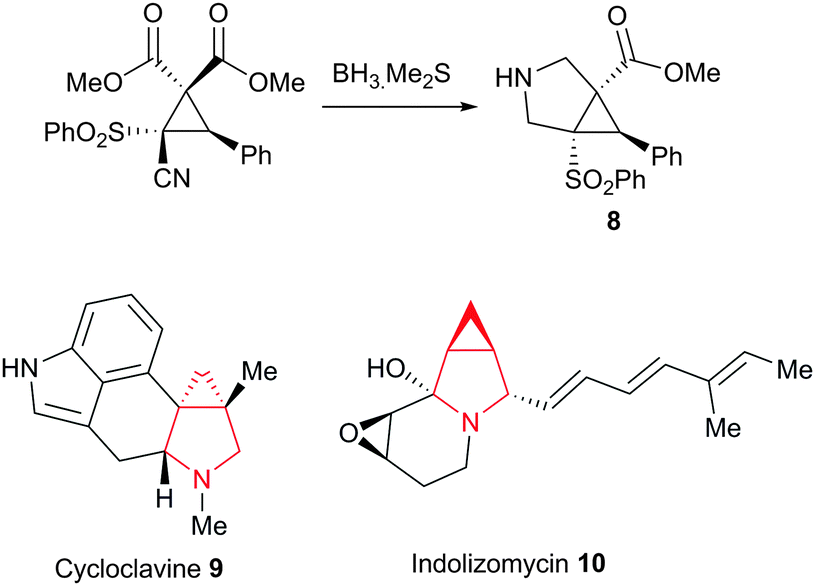 | ||
| Scheme 3 Synthesis of an intriguing 3-azabicyclo[3.1.0]hexane system. | ||
In conclusion, we have developed the enantioselective synthesis of a highly functionalised cyclopropane which can be transformed into several useful substrates. In particular, magnesium initiated radical desulfonylation-ring opening leads to the corresponding δ3-amino acid, and thus this route represents an extremely expedient way of accessing this class of molecule. In addition we have utilised the cyclopropane constructs to access a novel 3-azabicyclo[3.1.0]hexane system.
A.J.A.C. thanks GW Pharmaceuticals for funding (to L.S.A.), the Chemical Analysis Facility at the University of Reading, Dextra Laboratories for the use of high-pressure hydrogenation equipment, and the EPSRC UK National Mass Spectrometry Facility at Swansea University.
Notes and references
- For a review on the occurrences of cyclopropanes in nature and recent syntheses see D. Y.-K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631 RSC.
- M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804 RSC.
- (a) H. Pellissier, Tetrahedron, 2008, 64, 7041 CrossRef CAS PubMed; (b) G. Bartoli, G. Bencivenni and R. Dalpozzo, Synthesis, 2014, 979 Search PubMed; (c) H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed; (d) A. B. Charette and J.-F. Marcoux, Synlett, 1995, 1197 CrossRef CAS.
- (a) H. E. Simmons and R. D. Smith, J. Am. Chem. Soc., 1958, 80, 5323 CrossRef CAS; (b) H. E. Simmons and R. D. Smith, J. Am. Chem. Soc., 1959, 81, 4256 CrossRef CAS ; For catalytic asymmetric variations see; (c) H. Takahashi, M. Yoshioka, M. Ohno and S. Kobayashi, Tetrahedron Lett., 1992, 33, 2575 CrossRef CAS; (d) H. Shitama and T. Katsuki, Angew. Chem., Int. Ed., 2008, 47, 2450 CrossRef CAS PubMed; (e) H. Du, J. Long and Y. Shi, Org. Lett., 2006, 8, 2827 CrossRef CAS PubMed; (f) J. Long, Y. Yuan and Y. Shi, J. Am. Chem. Soc., 2003, 125, 13632 CrossRef CAS PubMed; (g) J. Long, H. Du, K. Li and Y. Shi, Tetrahedron Lett., 2005, 46, 2737 CrossRef CAS PubMed; (h) H. Y. Kim, A. E. Lurain, P. García- García, P. J. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2005, 127, 13138 CrossRef CAS PubMed; (i) R. G. Cornwall, O. A. Wong, H. Du, T. A. Ramirez and Y. Shi, Org. Biomol. Chem., 2012, 10, 5498 RSC.
- For recent discussion on this extensive area of catalysis, see (a) A. F. Trindade, J. A. S. Coelho, C. A. M. Afonso, L. F. Veiros and P. M. P. Gois, ACS Catal., 2012, 2, 370 CrossRef CAS; (b) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (c) H. M. L. Davies and J. R. Denton, Chem. Soc. Rev., 2009, 38, 3061 RSC and references therein.
- V. K. Aggarwal, E. Alonso, G. Fang, M. Ferrara, G. Hynd and M. Porcelloni, Angew. Chem., Int. Ed., 2001, 40, 1433 CrossRef CAS.
- (a) C. D. Papageorgiou, M. A. Cubillo de Dios, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2004, 43, 4641 CrossRef CAS PubMed; (b) C. C. C. Johansson, N. Bremeyer, S. V. Ley, D. R. Owen, S. C. Smith and M. J. Gaunt, Angew. Chem., Int. Ed., 2006, 45, 6024 CrossRef CAS PubMed.
- (a) R. K. Kunz and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 127, 3240 CrossRef PubMed ; For similar work see; (b) A. Hartikka and P. I. Arvidsson, J. Org. Chem., 2007, 72, 5874 CrossRef CAS PubMed; (c) A. Hartikka, A. T. Slosarczyka and P. I. Arvidsson, Tetrahedron: Asymmetry, 2007, 18, 1403 CrossRef CAS PubMed.
- See reference 3 and the appropriate section of C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2013, 114, 2390 CrossRef PubMed ; for detailed review.
- (a) A. J. A. Cobb, in Enantioselective Organocatalyzed Reactions II: Asymmetric C–C Bond Formation Processes, ed. R. Mahrwald, Springer, Dordrecht, 2011 Search PubMed; (b) L. S. Aitken, N. R. Arezki, A. Dell’Isola and A. J. A. Cobb, Synthesis, 2013, 2627 CAS.
- (a) S. Rajkumar, K. Shankland, J. M. Goodman and A. J. A. Cobb, Org. Lett., 2013, 15, 1386 CrossRef CAS PubMed . For a related process, see; (b) S. Meninno, G. Groce and A. Lattanzi, Org. Lett., 2013, 15, 3436 Search PubMed.
- For minireviews, see: (a) M. Kotke and P. R. Schreiner, in Hydrogen Bonding in Organic Synthesis, ed. P. Pihko, Wiley-VCH, 2009, pp. 141–351 Search PubMed; (b) H. Miyabe and Y. Takemoto, Bull. Chem. Soc. Jpn., 2008, 81, 785–795 CrossRef CAS; (c) S. J. Connon, Chem. Commun., 2008, 2499 RSC; (d) T. Marcelli, J. H. van Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed., 2006, 45, 7496 CrossRef CAS PubMed ; Other work by us in this area includes: ; (e) W. J. Nodes, D. R. Nutt, A. M. Chippindale and A. J. A. Cobb, J. Am. Chem. Soc., 2009, 131, 16016 CrossRef CAS PubMed; (f) S. Rajkumar, K. Shankland, G. D. Brown and A. J. A. Cobb, Chem. Sci., 2012, 3, 584 RSC.
- The C6–OH of cupreine has been shown to be very important in other reactions for obtaining high selectivities. See: (a) H. Li, Y. Wang, L. Tang and L. Deng, J. Am. Chem. Soc., 2004, 126, 9906 CrossRef CAS PubMed; (b) H. Li, Y. Wang, L. Tang, F. Wu, X. Liu, C. Guo, B. M. Foxman and L. Deng, Angew. Chem., Int. Ed., 2005, 44, 105 CrossRef CAS PubMed; (c) X. Liu, H. Li and L. Deng, Org. Lett., 2005, 7, 167 CrossRef CAS PubMed; (d) H. Li, J. Song, X. Liu and L. Deng, J. Am. Chem. Soc., 2005, 127, 8948 CrossRef CAS PubMed; (e) R. Dodda, J. J. Goldman, T. Mandal, C.-G. Zhao, G. A. Broker and E. R. T. Tiekink, Adv. Synth. Catal., 2008, 350, 537 CrossRef CAS PubMed; (f) P. Chauhan and S. S. Chimni, Tetrahedron Lett., 2013, 54, 4613 CrossRef CAS PubMed; (g) P. Chauhan and S. S. Chimni, RSC Adv., 2012, 2, 737 RSC.
- U. Das, Y.-L. Tsai and W. Lin, Org. Biomol. Chem., 2013, 11, 44 CAS.
- G. H. Lee, I. K. Youn, E. B. Choi, H. K. Lee, G. H. Yon, H. C. Yang and C. S. Pak, Curr. Org. Chem., 2004, 8, 1263 CrossRef CAS.
- The only previous report of this compound is in European Pat., EP1512678 A1, 2005, which obtained it in a 60% ee.
- For other (longer) syntheses, see (a) R. Karla, B. Ebert, C. Thorkildsen, C. Herdeis, T. N. Johansen, B. Nielsen and P. Krogsgaard-Larsen, J. Med. Chem., 1999, 42, 2053 CrossRef CAS PubMed; (b) Z.-T. He, Y.-B. Wei, H.-J. Yu, C.-Y. Sun, C.-G. Feng, P. Tian and G.-Q. Lin, Tetrahedron, 2012, 68, 9186 CrossRef CAS PubMed; (c) M. I. Attia, C. Herdeis and H. Bräuner-Osborne, Molecules, 2013, 18, 10266 CrossRef CAS PubMed (non-chiral).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1403683. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc05158d |
| This journal is © The Royal Society of Chemistry 2015 |