
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNear infrared light-driven water oxidation in a molecule-based artificial photosynthetic device using an upconversion nano-photosensitizer†
Xiaomin
Liu‡
a,
Hung-Cheng
Chen‡
b,
Xianggui
Kong
*a,
Youlin
Zhang
a,
Langping
Tu
a,
Yulei
Chang
a,
Fei
Wu
a,
Tongtong
Wang
a,
Joost N. H.
Reek
b,
Albert M.
Brouwer
 *b and
Hong
Zhang
*ab
*b and
Hong
Zhang
*ab
aState Key Laboratory of Luminescence and Applications, Changchun Institute of Optics, Fine Mechanics and Physics, Chinese Academy of Sciences, Changchun 130033, P. R. China. E-mail: xgkong14@ciomp.ac.cn
bVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH, Amsterdam. E-mail: h.zhang@uva.nl
First published on 2nd July 2015
Abstract
We provide the first demonstration of a near infrared light driven water oxidation reaction in a molecule-based artificial photosynthetic device using an upconversion nano-photosensitizer. One very attractive advantage of this system is that using NIR light irradiation does not cause significant photodamage, a serious problem in molecular based artificial photosynthesis under visible light irradiation.
Solar to fuel conversion using artificial photosynthetic devices has recently been considered to be a promising approach to produce sustainable energy.1,2 One of the options is solar driven water splitting by photoelectrochemical cells, resulting in the production of O2 and H2, which can be used as a clean fuel.3 The process consists of two half reactions: a proton reduction reaction at the photocathode and a water oxidation at the photoanode. Of these two half reactions, water oxidation is the more challenging process because it involves an endergonic four-electron process and therefore it often requires a large overpotential.4 In the photoanode design for photocatalytical water oxidation, effective catalyst and photoactive dye-sensitizer with broad-band light-harvesting function are needed to make this reaction as efficient as possible.5 Recently, we have successfully developed a novel photosensitizer Pt(II)-porphyrin (Pt(II)-TCPP) with a high potential of the radical cation/neutral redox couple that provides a driving force for favorable electron transfer between the catalyst and photo-oxidized Pt(II)-TCPP.6 Moreover, the photon capture ability is about three times higher than that of the widely used photosensitizer tris(2,2′-bipyridyl) ruthenium(II), [Ru(bpy)3]2+. Pt(II)-TCPP can utilize ultraviolet-visible light <555 nm for water-splitting when it is suitably coupled to an active water oxidation catalyst. In order to further improve the photon capture ability and potentially increase the solar to fuel conversion efficiency, the general design strategy is to introduce a cascade of events including excitation energy transfer by an artificial antenna using a multi-chromophore system.7–9 However, the bottleneck of solar driven water oxidation efficiency is still the limited absorption spectral range of the photosensitizer. The solution is to extend the absorption of the artificial photosynthetic system to longer wavelengths. In particular, the near-infrared (NIR) region contains ∼40% solar flux which usually remains unused.10
It is known that the light-harvesting units in photosystem II play a photo-protection role for avoiding photodamage by the safe thermal dissipation energy mechanisms under excess light.11–14 Likewise, improving the photostability is always a big concern in artificial photosynthetic device development for long-term activity.5 Although several mimicking models with the photo-protection function have been proposed, there is still a formidable challenge of further improving photocatalytic water oxidation.15–17 The advent of nanotechnology allows the integration of several photocatalytical elements in a nanoplatform18,19 and can also lead to new approaches to minimizing the photodamage effect.
Herein, we constructed NaYF4:Yb3+,Er3+ upconversion nanoparticles (UCNPs) covalently conjugated with our recently developed Pt(II)-TCPP photosensitizers. This nano-photosensitizer (UCNPs/Pt(II)-TCPP) can be excited with NIR light (∼920–980 nm) and utilizes the NIR photons to bring the Pt(II)-TCPP photosensitizer to its excited state. This is capable of driving photocatalytic water oxidation coupled to a Co4O4-cubane complex as water-oxidation catalyst in a neutral phosphate buffer solution (Fig. 1b).6 Furthermore, our nano-photosensitizer for NIR light driven water oxidation provides a novel design to prevent photodamage. For investigating the NIR-driven photocatalytic water oxidation activity, the conventional three component systems composed of a nano-photosensitizer, sacrificial electron acceptor and water oxidation catalyst were employed.20
 | ||
| Fig. 1 (a) Construction of the NaYF4:Yb3+,Er3+ UCNPs/Pt(II)-TCPP nano-photosensitizer. (b) Energy scheme of UCNPs/Pt(II)-TCPP and water oxidation catalyst. | ||
Hydrophobic NaYF4:20% Yb3+,0.2% Er3+ UCNPs were used as the energy donor. They are in the hexagonal phase and possess a uniform morphology with an average diameter of about 26 ± 2 nm (Fig. S1a and S2, ESI†). The hydrophilic NH2-functionalized UCNPs were then prepared via a ligand exchange process using poly(allylamine) as a surface coating agent. To ensure that the majority of Pt(II)-TCPP molecules were firmly linked to UCNPs, a covalent conjugation strategy was followed that involved a cross-linking reaction between the amino group of the UCNPs and the carboxyl group of the Pt(II)-TCPP, see Fig. 1a. Changes in the sizes of the nanoparticles before and after conjugation with Pt(II)-TCPP could not be deduced from the TEM images (Fig. S1a and b, ESI†) since the polymer coating is not readily observable in TEM images. In order to directly visualize the polymer shell, the sample was stained with phosphotungstic acid, which increases the contrast and makes the shells appear as halos surrounding the nanoparticles (Fig. S1c, ESI†). The polymer shell thickness was thus measured to be about 4.5 nm (Fig. S1d, ESI†). Coupling of the UCNPs with Pt(II)-TCPP was also confirmed by FTIR absorption spectra (Fig. S3, ESI†). The change in the carbonyl region (1650–1710 cm−1) indicates bond formation between the carboxylic acid group of Pt(II)-TCPP and the amino group of the nanoparticles. The loading capacity of Pt(II)-TCPP was then studied. Fig. S4 (ESI†) shows that the absorption intensities increased with the amount of added Pt(II)-TCPP, and saturated at 13%(w/w). Owing to the robust covalent bonding between Pt(II)-TCPP and NaYF4:Yb3+,Er3+, we could bind at most ∼2200 Pt(II)-TCPP molecules to each UCNP.
The design of this nano-photosensitizer is based on energy transfer from upconversion luminescent (UCL) states of UCNPs to photosensitizers. Effective energy transfer can be achieved by properly choosing a photosensitizer of which the absorption matches a desired UCL band of UCNPs and by shortening the interaction distance between the energy donor and the acceptor.21–23 Hybrid upconversion nano-photosensitizers have been successfully applied in different fields.24–26 Recently, improved solar water-splitting efficiency in semiconductor photoelectrode film incorporating UCNPs was also reported.27,28 In the present study, a photosensitizing molecule, Pt(II)-TCPP, was chosen because its absorption spectrum overlaps with the green UCL band (520–570 nm) (see Fig. 2a). Fig. 1b depicts the energy level scheme for photocatalytic components and reaction procedures. Firstly, under the excitation of 980 nm, the excited state Pt(II)-TCPP molecules are prepared via energy transfer from the green emission band of UCNPs. Subsequent highly exothermic triplet-state one-electron transfer reaction from photogenerated 3Pt(II)-TCPP to S2O82− in buffer solution results in the formation of the Pt(III)-porphyrin radical cation. This species with high reduction potential is thermodynamically capable of driving WOCs to oxidize water to O2. The energy transfer from UCNPs to Pt(II)-TCPP was confirmed from both steady-state UCL spectra and UCL decay kinetics. The UCL spectra of Fig. 2b demonstrate that under excitation at 980 nm, the 540 nm band was strongly quenched by Pt(II)-TCPP, while the 650 nm band remained unchanged. The energy transfer efficiency can be estimated from the quenching of UCL emission: E = (I0 − I1)/I0, where I0 and I1 are green emission intensities of UCNPs before and after conjugation with Pt(II)-TCPP, respectively. From this formula, the energy transfer efficiency of the 540 nm band was determined to be as high as 96.3% for the present covalently bonded nanoconjugates.
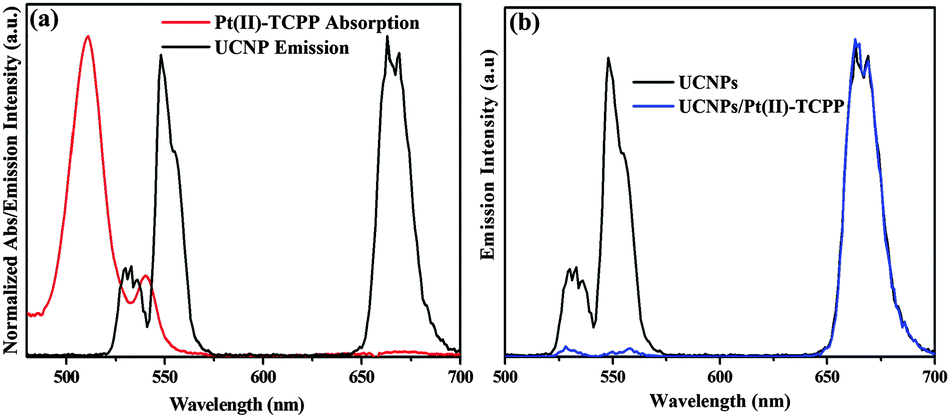 | ||
| Fig. 2 (a) Spectral overlap between the emission of the donor UCNPs and the absorption of the acceptor Pt(II)-TCPP in pH = 7.0, 0.1 M phosphate buffer. (b) Emission spectra of NH2-functionalized UCNPs and UCNPs/Pt(II)-TCPP nano-photosensitizer under excitation at 980 nm (normalized by the intensity at 650 nm). | ||
The dynamic energy transfer process, i.e. Förster resonant energy transfer (FRET), was studied by the temporal behavior of UCL recorded at 540 nm and 650 nm (Fig. S5, ESI†). In the presence of Pt(II)-TCPP, the average decay time decreases from ∼150 to ∼80 μs for 540 nm (Fig. S5a, ESI†), whereas the average decay time at 650 nm shows hardly any change due to the poor absorption of Pt(II)-TCPP in this region (Fig. S5b, ESI†). The dynamic energy transfer efficiency, calculated as (τ0 − τ1)/τ0, shows a value of 46.7% at 540 nm, which is less than the value determined from the steady-state UCL spectra (96.3%). This discrepancy indicates that re-absorption of Pt(II)-TCPP that is radiative and static in nature is non-negligible in the energy transfer process, i.e. both radiative and nonradiative energy transfer coexist in this case. However, the radiative energy transfer process does not alter the temporal behavior of UCL. For comparison we have also set up a model of radiative energy transfer by mixing UCNPs and Pt(II)-TCPP. The decay time at 540 nm shows almost no change (Fig. S5c, ESI†). In other words, the efficient energy transfer from UCNPs to Pt(II)-TCPP in the nano-photosensitizer ensures an effective photogeneration of excited Pt(II)-TCPP under NIR excitation.
Photocatalytic water oxidation experiments were carried out in solutions containing 1.83 × 10−3 M UCNPs/Pt(II)-TCPP nano-photosensitizer as evaluated by Pt(II)-TCPP absorption, 0.15 M Na2S2O8 and 1.5 × 10−4 M catalysts Co4O4-cubane in phosphate buffer solution (0.1 M, pH = 7.0) at room temperature. Photocatalytic oxygen generation was monitored through the detection of dissolved O2 using a Clark-type electrode. A 980 nm continuous diode laser was used as the irradiation source. The result of light driven oxygen formation is shown in Fig. 3a. Control experiments were carried out in which each individual component of the system was removed. Significant oxygen generation was observed only when all three components were present (Fig. S6, ESI†). A comparison was made between the covalently conjugated UCNPs/Pt(II)-TCPP nano-photosensitizer and the mixture as indicated in Fig. 3a. The maximum turnover frequency TOFmax of the catalyst was observed to be 6.6 × 10−4 mol O2 (mol of Co4O4-cubane)−1 s−1 in the nano-photosensitizer (excitation power 750 mW). However, the TOFmax was reduced to 9.6 × 10−5 mol O2 (mol of Co4O4-cubane)−1 s−1 in the mixture of UCNPs and Pt(II)-TCPP. These results indicate that efficient energy transfer in the covalently conjugated nano-photosensitizer significantly improves the light driven water oxidation activity, compared to the re-absorption process only in the mixture of UCNPs and Pt(II)-TCPP under the same excitation conditions. A further control experiment was conducted using Pt(II)-TCPP as the unique photosensitizer. Because Pt(II)-TCPP did not show any electronic absorption in the NIR range, there was, as expected, no oxygen evolution under the excitation of 980 nm light.
 | ||
| Fig. 3 (a) Photochemical oxygen evolution in 1.5 mL of a pH 7.0, 0.1 M phosphate buffer solution containing Co4O4 cubane (1.5 × 10−4 M), Na2S2O8 (0.15 M) and UCNPs/Pt(II)-TCPP nano-photosensitizer (1.83 × 10−3 M) (blue line); a mixture of UCNPs and Pt(II)-TCPP (black line). The red line is the control experiment with Pt(II)-TCPP only. Excitation was at 980 nm (750 mW). (b) Photochemical oxygen evolution with various excitation powers (980 nm laser) in 1.5 mL of a pH 7.0, 0.1 M phosphate buffer solution containing Na2S2O8 (0.15 M), UCNPs/Pt(II)-TCPP (1.83 × 10−3 M) and catalyst Co4O4 cubane (1.5 × 10−4 M). | ||
In order to investigate whether the TOF values are limited by the photon absorption rate or the inherent catalytic activity of the WOC, the light driven water oxidation activities were measured at different excitation powers (250 mW, 500 mW and 750 mW) of incident 980 nm laser light. The results are shown in Fig. 3b. The obtained values of TOFmax are 2.4 × 10−4, 4.1 × 10−4 and 6.6 × 10−4 mol O2 (mol of Co4O4-cubane)−1 s−1 under 250 mW, 500 mW and 750 mW, respectively, i.e. TOF increases with increasing excitation power. This result shows that NIR indirect excitation of Pt(II)-TCPP can keep up with the catalytic water oxidation cycle in this excitation power range. The overall quantum yield of NIR photons induced O2 generations is ∼9 × 10−3% in UCNPs/PtTCPP nanophotosensitizer under excitation with three different powers of 980 nm NIR-laser (for details of the calculation see the ESI†).
Photostability is a major concern for the performance of photosensitizers for photocatalytic water oxidation. UCNPs, which act as a nanotransducer to convert NIR light to visible wavelengths, afford an indirect, but local, light source which ensures that the excitation NIR light does not interact with any organic molecules in solution until it encounters the UCNPs/Pt(II)-TCPP nano-photosensitizer, different from the scenario directly using visible light irradiation. The photon energy can be effectively utilized since the surface of each UCNP is anchored with plenty of photosensitizers to accept the upconverted energy from the UCNPs, and thus can effectively avoid photodamage and greatly prolong the service life of the device. As shown in Fig. 4, 1.5 mL of a pH 7.0, 0.1 M phosphate buffer solution containing Na2S2O8 (0.15 M), UCNPs/Pt(III)-TCPP (1.83 × 10−3 M) and Co4O4-cubane (1.5 × 10−4 M) was irradiated under 980 nm laser (indirect irradiation) and 532 nm laser (direct irradiation) for comparison. The power of the laser was 300 mW for 980 nm and 100 mW for 532 nm, respectively, which led to almost equal oxygen generation rates (Fig. 4a). After irradiation for 20 min, UV-Vis absorption spectra were recorded to evaluate the photostability, as shown in Fig. 4b. The UV-Vis absorption spectrum of the sample after 980 nm irradiation did not show any change. In contrast, the UV-Vis absorption spectrum of the sample was completely changed upon 532 nm irradiation, indicating severe photodamage under direct excitation. The photos of the samples before and after light irradiation of the two different wavelengths (inset in Fig. 4b and Fig. S7, ESI†) provide a vivid illustration of the difference in photostability under different conditions.
 | ||
| Fig. 4 (a) Photochemical oxygen evolution in 1.5 mL of a pH 7.0, 0.1 M phosphate buffer solution with different excitation powers of 980 nm laser (300 mW, blue) and 532 nm green laser (100 mW, red). (b) Absorption spectra of samples before irradiation, after 980 nm irradiation and after 532 nm irradiation. The inset in the figure shows the photos of the samples before and after irradiations. | ||
To understand this phenomenon we need to go into the working mechanism of the photosynthetic device. The energy level diagram is shown in Fig. 1b including combinations of water oxidation catalyst, nano-photosensitizer and sacrificial electron acceptor (Na2S2O8) in neutral phosphate buffer. Under the excitation of 980 nm, Pt(III)-TCPP molecules are prepared in the excited state via energy transfer from UCNPs. Highly exothermic electron transfer from photogenerated 3Pt(II)-TCPP to S2O82− in buffer solution results in the formation of Pt(II)-porphyrin radical cations. This species is thermodynamically capable of driving the water oxidation catalyst to oxidize water to O2.6 The UCNPs/Pt(II)-TCPP nano-photosensitizer solution without the addition of sacrificial electron acceptor (sodium persulfate) retains a similar absorption spectra under light irradiation of both excitation wavelengths (see Fig. S8, ESI†). This indicates that the photodamage paths are mostly related to the radical cations of Pt(II)-TCPP. The reduction of the radical cation of the photosensitizer by the water oxidation catalyst is rate limiting in general.29 Therefore side reactions of the radical cations may be relatively enhanced at high incident light power The porphyrin radical cation in water shows a broad and intense absorption in the UV-Visible range and has a lifetime that can be up to seconds.30,31 Visible light (e.g. 532 nm) may cause photochemical degradation via the excited state of the metalloporphyrin radical cation. Conversely, photo-absorption of porphyrin radical cation can be excluded at 980 nm. Since most of the upconverted 540 nm band contributes to bringing the photosensitizers to the excited states due to the efficient energy transfer between the UCNPs and photosensitizers, the residual upconverted 540 nm band is much weaker than the direct 532 nm laser irradiation for the photodamage of the radical cations, which might be responsible for the significantly enhanced photostability of UCNPs/Pt(II)-TCPP nano-photosensitizers under NIR excitation.
In conclusion, we have demonstrated a novel NIR driven nano-photosensitizer, integrating UCNPs with Pt(II)-TCPP. The excited state of this sensitizer has sufficient oxidizing power to drive the water splitting reaction when an appropriate catalyst is present. To the best of our knowledge, this is the first demonstration of a light driven water oxidation reaction using only near-infrared photons in a molecule-based artificial photosynthetic device. Furthermore, the UCNPs/Pt(II)-TCPP nano-photosensitizer also significantly improves the photostability of the solution under NIR light excitation compared to the scenario of visible light excitation. The upconversion efficiency should be further improved for practical applications in actual solar driven catalysis. This can be achieved by manipulating emission and excitation processes through structure design or plasmonic coupling.32,33 Consequently, harvesting NIR photons for light driven water oxidation by artificial photosynthesis can be achieved under solar irradiation.
Notes and references
- S. Bensaid, G. Centi, E. Garrone, S. Perathoner and G. Saracco, ChemSusChem, 2012, 5, 500 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724 CrossRef CAS PubMed.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357 RSC.
- H. C. Chen, G. H Hetterscheid, R. M. Williams, J. I. Van der Vlugt, J. N. H. Reek and A. M. Brouwer, Energy Environ. Sci., 2015, 8, 975 CAS.
- H. J. Imahori, J. Phys. Chem. B, 2004, 108, 6130 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS PubMed.
- F. Puntoriero, G. Ganga, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and S. Campagna, Chem. Commun., 2010, 46, 4725 RSC.
- N. D. McDaniel and S. Bernhard, Dalton Trans., 2010, 39, 10021 RSC.
- J. Barber and B. Andersson, Trends Biochem. Sci., 1992, 17, 61 CrossRef CAS.
- H. A. Frank and G. W. Brudvig, Biochemistry, 2004, 43, 8607 CrossRef CAS PubMed.
- N. E. Holt, G. R. Fleming and K. K. Niyogi, Biochemistry, 2004, 43, 8281 CrossRef CAS PubMed.
- G. Scholes, G. Fleming, A. Olaya-Castro and R. Van Grondelle, Nat. Chem., 2011, 3, 763 CrossRef CAS PubMed.
- R. Berera, C. Herrero, I. H. M. Van Stokkum, M. Vengris, G. Kodis, R. E. Palacios, H. Van Amerongen, R. Van Grondelle, D. Gust and T. A. Moore, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5343 CrossRef CAS PubMed.
- S. D. Straight, G. Kodis, Y. Terazono, M. Hambourger, T. A. Moore, A. L. Moore and D. Gust, Nat. Nanotechnol., 2008, 3, 280 CrossRef CAS PubMed.
- Y. Terazono, G. Kodis, K. Bhushan, J. Zaks, C. Madden, A. L. Moore, G. R. Fleming and D. Gust, J. Am. Chem. Soc., 2011, 133, 2916 CrossRef CAS PubMed.
- G. Yuan, A. Agiral, N. Pellet, W. Kim and H. Frei, Faraday Discuss., 2014, 176, 233 RSC.
- T. Itoh, K. Yano, T. Kajino, S. Itoh, Y. Shibata, H. Mino, R. Miyamoto, Y. Inada, S. Iwai and Y. Fukushima, J. Phys. Chem. B, 2004, 108, 13683 CrossRef CAS.
- L. Duan, L. Tong, Y. Xu and L. Sun, Energy Environ. Sci., 2011, 4, 3296 CAS.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161 CrossRef CAS PubMed.
- X. Han, R. Deng, X. Xie and X. Liu, Angew. Chem., Int. Ed., 2014, 53, 11702 CrossRef PubMed.
- J. Liu, Y. Liu, Q. Liu, C. Li, L. Sun and F. Li, J. Am. Chem. Soc., 2011, 133, 15276 CrossRef CAS PubMed.
- T. Q. Wu, J. C. Boyer, M. Barker, D. Wilson and N. R. Branda, Chem. Mater., 2013, 25, 2495 CrossRef CAS.
- X. Lu, X. G. Kong, X. M. Liu, L. P. Tu, Y. L. Zhang, Y. L. Chang, K. Liu, D. Z. Shen, H. Y. Zhao and H. Zhang, Biomaterials, 2014, 35, 4146 CrossRef PubMed.
- N. M. Idris, M. K. Gnanasammandhan, J. Zhang, P. C. Ho, R. Mahendran and Y. Zhang, Nat. Med., 2012, 18, 1580 CrossRef CAS PubMed.
- M. Zhang, Y. Lin, T. J. Mullen, W. F. Lin, L. D. Sun, C. H. Yan, T. E. Patten, D. Wang and G. Y. Liu, J. Phys. Chem. Lett., 2012, 3, 3188 CrossRef CAS.
- F. Gonell, M. Haro, R. S. Sa, P. Negro and B. Julia, J. Phys. Chem. C, 2014, 118, 11279 CAS.
- W. J. Youngblood, S. H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926 CrossRef CAS PubMed.
- A. Harriman, P. Neta and M. C. Richoux, J. Phys. Chem., 1986, 90, 3444 CrossRef CAS.
- A. Harriman, G. Porter and P. Walters, J. Chem. Soc., Faraday Trans., 1983, 79, 1335 RSC.
- W. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov and J. C. Hummelen, Nat. Photonics, 2012, 6, 560 CrossRef CAS PubMed.
- D. M. Wu, A. García-Etxarri, A. Salleo and J. A. Dionne, J. Phys. Chem. Lett., 2014, 5, 4020 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and additional data. See DOI: 10.1039/c5cc05102a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |