
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn optimized polyamine moiety boosts the potency of human type II topoisomerase poisons as quantified by comparative analysis centered on the clinical candidate F14512†
Giulia
Palermo
a,
Elirosa
Minniti
ac,
Maria Laura
Greco
b,
Laura
Riccardi
a,
Elena
Simoni
c,
Marino
Convertino
a,
Chiara
Marchetti
c,
Michela
Rosini
c,
Claudia
Sissi
b,
Anna
Minarini
c and
Marco
De Vivo
*ad
aLaboratory of Molecular Modeling and Drug Discovery, Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genoa, Italy. E-mail: marco.devivo@iit.it
bDepartment of Pharmaceutical and Pharmacological Sciences, University of Padova, Via Marzolo 5, 35131 Padova, Italy
cDepartment of Pharmacy and Biotechnology, Alma Mater Studiorum-University of Bologna, Via Belmeloro 6, 40126 Bologna, Italy
dIAS-5/INM-9 Computational Biomedicine Forschungszentrum Jülich, Wilhelm-Johnen-Straße, 52428 Jülich, Germany
First published on 27th July 2015
Abstract
Combined computational–experimental analyses explain and quantify the spermine-vectorized F14512's boosted potency as a topoII poison. We found that an optimized polyamine moiety boosts drug binding to the topoII/DNA cleavage complex, rather than to the DNA alone. These results provide new structural bases and key reference data for designing new human topoII poisons.
Topoisomerase–targeted drugs are considered poisons when they act by trapping the covalent enzyme/DNA cleavage complex, which is formed during the catalytic cycle required for DNA topology modification.1–6 The spermine-vectorized F14512 is one of the most promising anticancer agents currently in clinical trials for the treatment of refractory/relapsing acute myeloid leukemia (AML).7,8 Gentry et al.9 recently reported that it acts as a poison of human type II topoisomerase (topoII), much like its parent anticancer drug etoposide, which bears a glycosidic moiety at C4 in place of the spermine (Fig. 1).9–11 Remarkably, F14512 is reported to be ∼10-fold more potent than etoposide in inhibiting cell proliferation.11 This is partly attributed to the spermine-mediated F14512 uptake by the polyamine transport system (PTS), which is overactive in many tumor cells.7,11 However, the conserved epipodophyllotoxin core and the mechanism of action suggest that the enhanced efficacy of F14512 in comparison to that of etoposide might also come from favourable interactions of its spermine moiety within the topoII/DNA cleavage complex.9 This is still uncertain, since nobody has yet reported an atomic-level description and evaluation of the interaction between the spermine-conjugate F14512 and the topoII/DNA cleavage complex.
 | ||
| Fig. 1 (A) Model of the ternary F14512/topoII/DNA cleavage complex; (B) chemical structures of compounds 1–7. | ||
Here, we used molecular modeling and extensive simulations to identify the most probable configurations of the ternary F14512/topoII/DNA cleavage complex (F14cc), which best correspond to the existing structural and NMR spectroscopy data.9,12 F14512 was initially docked to the binary topoII/DNA cleavage complex using a positional restraint grounded on the underlying assumption of an etoposide-like binding mode for the conserved epipodophyllotoxin core.13 For comparison, we simulated a model system of the ternary etoposide/topoII/DNA complex (ETOcc), based on the recent crystal structure of Wu et al.12 Then, we performed a comparative molecular dynamics (MD) analysis of F14cc and ETOcc, on a total of ∼350–400 ns of classical MD per system.14
We identified system-dependent properties and key interactions between the topoII/DNA receptor and the drug, either F14512 or etoposide. The conserved E-ring of the two drugs is equally stable in the two systems (RMSD ∼0.35/0.41 ± 0.02 Å, Fig. 2 and Fig. S2, ESI†), due to a highly conserved H-bond with Asp479 – preserved for 82.6% and 78.7% of the simulation time in ETOcc and F14cc, respectively (Fig. S3, Movies S1 and S2, ESI†). In this position, the E-ring likely favours a large perturbation of the catalytic two-metal-ion coordination sphere in topoII, blocking the topoII-mediated DNA religation step.1,12,15–22
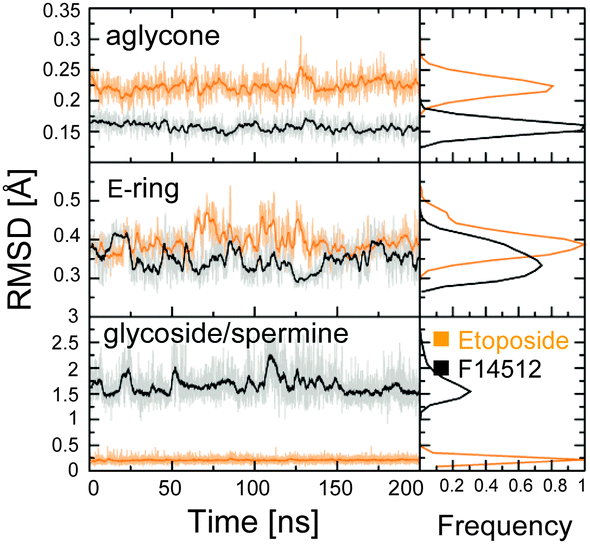 | ||
| Fig. 2 Time evolution (last 200 ns) of the RMSD for the heavy atoms of the aglycone core (upper graph), the E-ring (central graph), and the glycoside/spermine group (lower panel) of etoposide (orange) and F14512 (black) in the ETOcc and F14cc systems, for one subunit of topoII. Full data in Fig. S2 (ESI†). | ||
The aglycone core is also very stable in both systems (RMSD of ∼0.16 ± 0.02 Å and ∼0.23 ± 0.02 Å in F14cc and ETOcc, respectively). F14512 maintains a firm interaction, conserved for ∼96.6% of the simulation time (Fig. S3, ESI†), between its amide nitrogen (N1, Fig. 3) and the DNA base G+5. Although the aglycone core remains between the T+1/A+4 and C−1/G+5 DNA bases in both F14cc and ETOcc systems, it does not form typical stacking interactions with the T+1/A+4 and C−1/G+5 base pairs. This is due to its conserved tilted orientation relative to the DNA backbones and the local widening of the cleaved DNA strand. This non-intercalative mode for drug binding to DNA (Movie S2, ESI†) agrees with previous findings of structural and biophysical studies.9,12
 | ||
| Fig. 3 Key interactions between F14512 and the topoII/DNA cleavage complex shown schematically (top). The statistical distribution over the production runs of the H-bond interactions is reported in Fig. S3 (ESI†). Three representative binding modes of F14512 from MD simulations are shown with different colours of the spermine moiety (bottom). | ||
Interestingly, during our MD simulations, we observed a key conformational mobility for the F14512 spermine tail, which shows an RMSD of ∼1.88 ± 0.36 Å (Fig. 2 and Fig. S2, ESI†). The long polyamine chain extends toward the major groove and interacts with the backbone phosphates of both DNA strands (Fig. 3). In detail, the amine nitrogen atoms in positions 13′′ and 17′′ (N13′′ and N17′′) of the spermine tail act as key anchors for binding. The N13′′ atom interacts mainly with the DNA phosphates of C+6 and C−6, while N17′′ interacts mainly with C+6 and G−7. The spermine nitrogen N17′′ alternatively H-bonds the protein residues Glu953 and Glu519, which are located close to the substrate DNA.
These transient interactions occur at both DNA strands with comparable statistical distributions (Fig. S3, ESI†). The amine nitrogen in position 8′′ (N8′′) of the spermine chain provides additional DNA anchoring, establishing interactions for ∼36% of the simulation time with G+5. Lastly, the amine nitrogen in position 4′′ (N4′′) contributes only marginally to stabilizing the drug to the cleavage complex, being mostly oriented toward the solvent during the simulations. This complex H-bond network reflects a favourable complementarity of F14512 and the topoII/DNA cleavage complex. Etoposide, in contrast, cannot form such an H-bond network. Its glycosidic moiety at C4 is highly stable (RMSD of ∼0.21 ± 0.05 Å) and interacts with the G+5 carbonyl for ∼71.4% of the simulation time (Fig. S3, ESI†), protruding toward the DNA major groove and remaining stably located near Gln778 and Met782. Finally, there are similar hydrophobic contacts of the drug with the protein in both ETOcc and F14cc (Fig. S4, ESI†).
Thus, the tighter drug binding of F14512 seems to be mainly due to the spermine chain, which forms, through the major groove, extensive favourable drug–target interactions with both DNA and topoII. The stronger interaction of F14512 with the targeted cleavage complex is also confirmed through steered MD simulations (Fig. 4, full details in the ESI†).23 Although only qualitative, the calculated unbinding force profiles and the external work for the undocking of the two compounds from the target display tighter binding for F14512 (∼80 kcal mol−1 more work needed for the unbinding of F14512 from the cleavage complex, compared to etoposide). Again, this is due to the numerous H-bond interactions formed between the spermine tail and the cleavage complex, which need to be disrupted during the undocking dynamics (Movie S3, ESI†).23
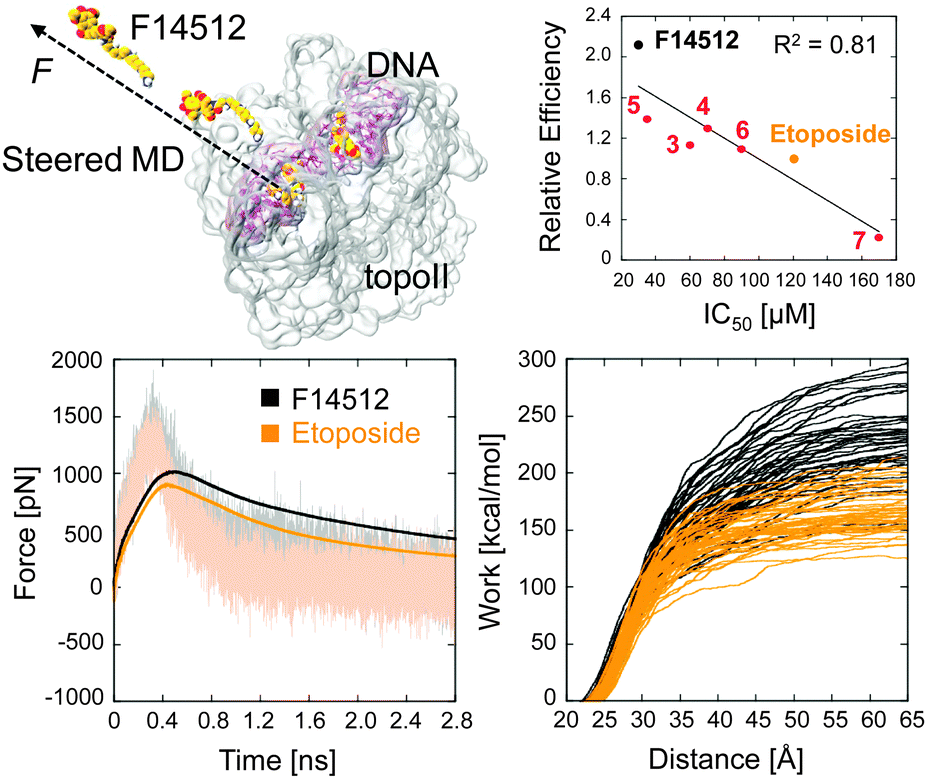 | ||
| Fig. 4 Low row: Average unbinding force profiles (left) and external work (right) for the undocking of F14512 and etoposide from the cleavage complex, as calculated from multiple steered MD simulations (top-left). Full details in the ESI.† Top-right: Measured relative efficiencies vs. IC50 values for compounds 1–7 (see Table 1). | ||
To validate this computational evidence for a key role of the polyamine chain in boosting the binding of F14512 to the cleavage complex, we designed and synthesized five new polyamine-conjugates 3–7 (see the ESI† for chemical synthesis). These are the spermidine derivative 3, the methyl spermine derivative 4 and compounds 5 and 6, which bear a spermine analogue chain with a shorter methylene spacer (two and three methylenes, respectively) between the two inner nitrogen atoms. In 7, the inner nitrogen atoms of the spermine portion of F14512 were replaced with oxygens to explore the importance of the polycationic chain in increasing topoII inhibition (Fig. 1). Compounds 1–7 were tested against human topoII. The IC50 values obtained in a relaxation inhibition assay, summarized in Table 1, confirm F14512 as the most potent derivative in the series, with an IC50 of ∼30 μM, which is ∼4-fold better than etoposide in the same experimental assay.
| Compound | IC50a [μM] | Relative efficiencyb | % Abs changec |
|---|---|---|---|
| a Compound concentration required to inhibit the relaxation activity of topoII (IC50). b Extent of cleavage product formation in comparison to etoposide (relative efficiency). c Variation of the absorbance signal at 290 nm induced by the addition of four equivalent of ctDNA (% Abs change). | |||
| Etoposide | 120 ± 10 | 1 | nd |
| F14512 | 30 ± 5 | 2.12 ± 0.22 | 17.7 ± 1.0 |
| 3 | 60 ± 8 | 1.13 ± 0.19 | 7.7 ± 0.7 |
| 4 | 70 ± 19 | 1.30 ± 0.03 | 9.5 ± 1.7 |
| 5 | 35 ± 4 | 1.39 ± 0.01 | 11.5 ± 4.0 |
| 6 | 90 ± 5 | 1.09 ± 0.26 | 6.7 ± 5.0 |
| 7 | 170 ± 20 | 0.22 ± 0.01 | 13.0 ± 2.4 |
Additionally, all the tested derivatives stabilized the topoII/DNA cleavage complex (Table 1) thus indicating that all of them are topoII poisons. The amount of cleaved DNA produced by the enzyme in the presence of each polyamine derivative was quantified and compared to the one produced in the presence of etoposide at the same concentration (5–50 μM concentration range). The ratio of these values provided the “relative efficiency” reported in Table 1. Although only indicative, an excellent linear correlation (R2 = 0.81, Fig. 4) was found between the observed relaxation activity of topoII and the extent of cleavage complex formation. This result indicates that the impairment of the enzymatic activity generated by all the examined compounds occurs according to an overall shared mechanism of action. Interestingly, the relative potency of these inhibitors strictly depends on the structural features of the polyamine chain. The activity is reduced by ∼2-fold by the replacement of the tetramine spermine with the triamine spermidine (3) or the transformation of the terminal primary amine into a secondary amine (4).
The low inhibition can be explained by the fact that a shorter polyamine chain, as in 3 and 6, does not allow engagement of distant topoII residues such as Glu519 and Glu953. Compound 4, with a methyl spermine tail, is unable to form an optimal interaction between its terminal secondary amine and the carboxylate groups of Glu519 and Glu953, due to the steric hindrance of the methyl group (Fig. S10, ESI†). These drug–topoII interactions are instead observed with F14512 in MD simulations (Fig. 3). Finally, 7 was the weakest and least efficient inhibitor of topoII (∼170 μM, Table 1) of the series, proving the key role of the polycationic character of the chain in drug activity.
As evidenced by previous structural data of etoposide in complex with the binary topoII/DNA complex,12 a few contacts are formed between the C4-substituent and topoII. In fact, in the absence of DNA, the drug shows negligible interactions with topoII, alone.9,24 These results demonstrated that the drug has limited, if any, interaction with topoII, alone, while it binds tightly to the topoII/DNA cleavage complex. Here, we also quantified the efficiency of our ligands to bind the DNA, alone (i.e., in the absence of topoII), via UV measurements. In Table 1 we report the variation of the absorbance signal induced by DNA, which reflects the extent of the bound ligand to the DNA. The results indicate that all polyamine conjugates were able to bind DNA, as previously reported for F14512,11 which here emerges as the strongest DNA binder. Notably, 7 is also a good DNA binder, which suggests that the central amines of the polyamine chain are critical for drug binding to the topoII/DNA cleavage complex, rather than to the DNA alone.
Docking calculations further support the evidence that the most active topoII poisons are those that, through the polyamine chain, form an extended network of H-bonds within the topoII/DNA cleavage complex. In fact, the score distribution of the obtained poses for F14512 and compounds 3–7 docked to the topoII/DNA cleavage complex reproduces well the IC50 and relative efficiency trend values (full details in the ESI† and Fig. S5–S7). The docking ensemble of F14512 shows the spermine, which extends into the major groove, interacting with both DNA strands, as in MD, whereas the tail of 7, although of the same length, is mostly located far from the DNA backbone, assuming curved conformations that cannot form stable and favourable interactions with the targeted complex (Fig. S5–S7, ESI†). This underlines that the polyamine moiety's favourable contribution is not simply connected to the efficiency of DNA recognition but is due to the stabilization of the topoII/DNA cleavage complex.
In summary, our study unravels crucial drug–target configurations of the still structurally uncharacterized F14512/topoII/DNA complex that best correspond to experimental results.9,11,25,26 Extensive MD simulations suggest key drug–target interactions that explain F14512's boosted potency as a topoII poison. Our computational evidence is then validated through experiments that, importantly, also demonstrate that an optimized polyamine moiety boosts drug binding to the topoII/DNA cleavage complex, rather than to the DNA alone. This is shown here for the spermine-vectorized F14512, currently in clinical trials, and other similar polyamine-conjugated derivatives of epipodophyllotoxin. Taken together, these results offer an additional structural basis and key reference data for designing and assessing novel human topoII poisons for anticancer drug discovery.5,27–30
This work has been supported in part by the Italian Association for Cancer Research (AIRC) through the “MFAG n. 14140” Grant and by Università degli Studi di Padova (Grant # 60A04-7255). MLG was funded by CARIPARO. We also thank PRACE for HPC computing time. GP thanks Dr Anna Berteotti for useful discussions. We thank Grace Fox for proofreading the manuscript.
Notes and references
- J. E. Deweese and N. Osheroff, Nucleic Acids Res., 2009, 37, 738–748 CrossRef CAS PubMed.
- W. Yang, Crit. Rev. Biochem. Mol. Biol., 2010, 45, 520–534 CrossRef CAS PubMed.
- B. Pogorelcnik, A. Perdih and T. Solmajer, Curr. Pharm. Des., 2013, 19, 2474–2488 CrossRef CAS.
- J. E. Deweese, M. A. Osheroff and N. Osheroff, Biochem. Mol. Biol. Educ., 2008, 37, 2–10 CrossRef PubMed.
- Y. Pommier, E. Leo, H. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS PubMed.
- Y. Pommier, ACS Chem. Biol., 2013, 8, 82–95 CrossRef CAS PubMed.
- A. Kruczynski, A. Pillon, L. Creancier, I. Vandenberghe, B. Gomes, V. Brel, E. Fournier, J. P. Annereau, E. Currie, Y. Guminski, D. Bonnet, C. Bailly and N. Guilbaud, Leukemia, 2013, 27, 2139–2148 CrossRef CAS PubMed.
- F. Mouawad, A. Gros, B. Rysman, C. Bal-Mahieu, C. Bertheau, S. Horn, T. Sarrazin, E. Lartigau, D. Chevalier, C. Bailly, A. Lansiaux and S. Meignan, Oral Oncol., 2014, 50, 113–119 CrossRef CAS PubMed.
- A. C. Gentry, S. L. Pitts, M. J. Jablonsky, C. Bailly, D. E. Graves and N. Osheroff, Biochemistry, 2011, 50, 3240–3249 CrossRef CAS PubMed.
- C. Bailly, Chem. Rev., 2012, 112, 3611–3640 CrossRef CAS PubMed.
- J. M. Barret, A. Kruczynski, S. Vispe, J. P. Annereau, V. Brel, Y. Guminski, J. G. Delcros, A. Lansiaux, N. Guilbaud, T. Imbert and C. Bailly, Cancer Res., 2008, 68, 9845–9853 CrossRef CAS PubMed.
- C. C. Wu, T. K. Li, L. Farh, L. Y. Lin, T. S. Lin, Y. J. Yu, T. J. Yen, C. W. Chiang and N. L. Chan, Science, 2011, 333, 459–462 CrossRef CAS PubMed.
- Docking (Glide) and MD simulations (NAMD 2.8) were performed using the pdb 3QX3 (solved at 2.16 Å resolution – see ref. 12). The amine groups of the polyamine chains were protonated as predicted by the Epik software, at pH 7.4 (see Fig. 3, and the ESI†).
- Both ETOcc and F14cc were immersed in a box of TIP3P waters, for a total size of ∼220
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms, each. The AMBER force field ff99Bildn3 was adopted for the protein, whereas parmbsc04 was adopted for the DNA. Etoposide and F14512 were treated with GAFF and RESP charges. Monomers of topoII systems were considered separately for comparative analysis, thus doubling the statistics. See the ESI† for full details.
000 atoms, each. The AMBER force field ff99Bildn3 was adopted for the protein, whereas parmbsc04 was adopted for the DNA. Etoposide and F14512 were treated with GAFF and RESP charges. Monomers of topoII systems were considered separately for comparative analysis, thus doubling the statistics. See the ESI† for full details. - J. E. Deweese, A. M. Burch, A. B. Burgin and N. Osheroff, Biochemistry, 2009, 48, 1862–1869 CrossRef CAS PubMed.
- J. E. Deweese, A. B. Burgin and N. Osheroff, Nucleic Acids Res., 2008, 36, 4883–4893 CrossRef CAS PubMed.
- J. E. Deweese, F. P. Guengerich, A. B. Burgin and N. Osheroff, Biochemistry, 2009, 48, 8940–8947 CrossRef CAS PubMed.
- J. E. Deweese and N. Osheroff, Metallomics, 2010, 2, 450–459 RSC.
- G. Palermo, A. Cavalli, M. L. Klein, M. Alfonso-Prieto, M. Dal Peraro and M. De Vivo, Acc. Chem. Res., 2015, 48, 220–228 CrossRef CAS PubMed.
- G. Palermo, M. Stenta, A. Cavalli, M. Dal Peraro and M. De Vivo, J. Chem. Theory Comput., 2013, 9(2), 857–862 CrossRef CAS.
- C. Sissi and M. Palumbo, Nucleic Acids Res., 2009, 37, 702–711 CrossRef CAS PubMed.
- B. H. Schmidt, A. B. Burgin, J. E. Deweese, N. Osheroff and J. M. Berger, Nature, 2010, 465, 641–644 CrossRef CAS PubMed.
- F. Colizzi, R. Perozzo, L. Scapozza, M. Recanatini and A. Cavalli, J. Am. Chem. Soc., 2010, 132, 7361–7371 CrossRef CAS PubMed.
- R. P. Bender, M. J. Jablonksy, M. Shadid, I. Romaine, N. Dunlap, C. Anklin, D. E. Graves and N. Osheroff, Biochemistry, 2008, 47, 4501–4509 CrossRef CAS PubMed.
- I. Laponogov, X. S. Pan, D. A. Veselkov, K. E. McAuley, L. M. Fisher and M. R. Sanderson, PLoS One, 2010, 5, e11338 Search PubMed.
- I. Laponogov, M. K. Sohi, D. A. Veselkov, X. S. Pan, R. Sawhney, A. W. Thompson, K. E. McAuley, L. M. Fisher and M. R. Sanderson, Nat. Struct. Mol. Biol., 2009, 16, 667–669 CAS.
- M. De Vivo, Front. Biosci., Landmark Ed., 2011, 16, 1619–1633 CrossRef CAS PubMed.
- M. Nagarajan, X. Xiao, S. Antony, G. Kohlhagen, Y. Pommier and M. Cushman, J. Med. Chem., 2003, 46, 5712–5724 CrossRef CAS PubMed.
- C. C. Wu, Y. C. Li, Y. R. Wang, T. K. Li and N. L. Chan, Nucleic Acids Res., 2013, 41, 10630–10640 CrossRef CAS PubMed.
- M. N. Drwal, J. Marinello, S. G. Manzo, L. P. Wakelin, G. Capranico and R. Griffith, PLoS One, 2014, 9, e114904 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc05065k |
| This journal is © The Royal Society of Chemistry 2015 |