
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A recyclable CO surrogate in regioselective alkoxycarbonylation of alkenes: indirect use of carbon dioxide†
P. H.
Gehrtz
,
V.
Hirschbeck
and
I.
Fleischer
*
Institute of Organic Chemistry, University of Regensburg, Universitätsstraße 31, 93040 Regensburg, Germany. E-mail: ivana.fleischer@chemie.uni-regensburg.de
First published on 2nd July 2015
Abstract
Herein, we report a Pd-catalysed alkoxycarbonylation of alkenes based on the use of a recyclable CO2 reduction product, the crystalline and air-stable N-formylsaccharin, as a CO surrogate. The carbonylation proceeds under ambient conditions in an exceptionally complementary regioselective fashion yielding the desired branched products from styrene derivatives and valuable linear esters from alkyl-substituted alkenes.
Carbon monoxide constitutes the most versatile C1 building block for the construction of carbonyl compounds in homogeneously catalysed reactions.1 Carbonylations of alkenes, such as hydroformylation2 and alkoxycarbonylation3 play a central role in the production of bulk chemicals. However, their use in small laboratory synthetic applications is more rare, because of the toxicity of CO and advanced technical requirements. Therefore, the development of alternative ways to produce stoichiometrical amounts of CO in the reaction mixture is of considerable interest in synthetic organic chemistry.4
Thus, a number of CO surrogates have been applied in the carbonylation reactions of alkenes over the years. Most recent developments include the use of formates,5 formic acid,6 formaldehyde,7 alcohols8 and the greenhouse gas carbon dioxide.9 However, most transformations are performed under forcing conditions (temperatures >100 °C, high catalyst loading) and even if cheap, low-weight carbonylation reagents are used, the overall atom economy is deteriorated due to the employment of overstoichiometrical amounts of the CO surrogate. Moreover, the use of an internal alternative CO source often leads to deviations from the carbonylation mechanism causing changes in reactivity and selectivity.
In order to overcome the flaws of the known CO surrogates used in alkene carbonylations, we have developed a methodology based on a separate CO production originally reported by Skrydstrup for the carbonylations of ArX compounds.10 Palladium-catalysed alkoxycarbonylation11 of styrene derivatives was chosen as a model reaction and the catalyst was optimized in order to operate under mild conditions and selectively produce branched esters. Thus, a unique catalytic system was developed, which is able to carbonylate alkenes at the room temperature with only 0.5 mol% catalyst loading and utilize N-formylsaccharin as a recyclable CO surrogate (Scheme 1).12 Notably, our strategy allows for an indirect use of carbon dioxide as a C1 source. This approach to the reductive activation of CO2 in two steps constitutes a useful alternative to the literature known methods, which usually suffer from harsh reaction conditions and low selectivity.
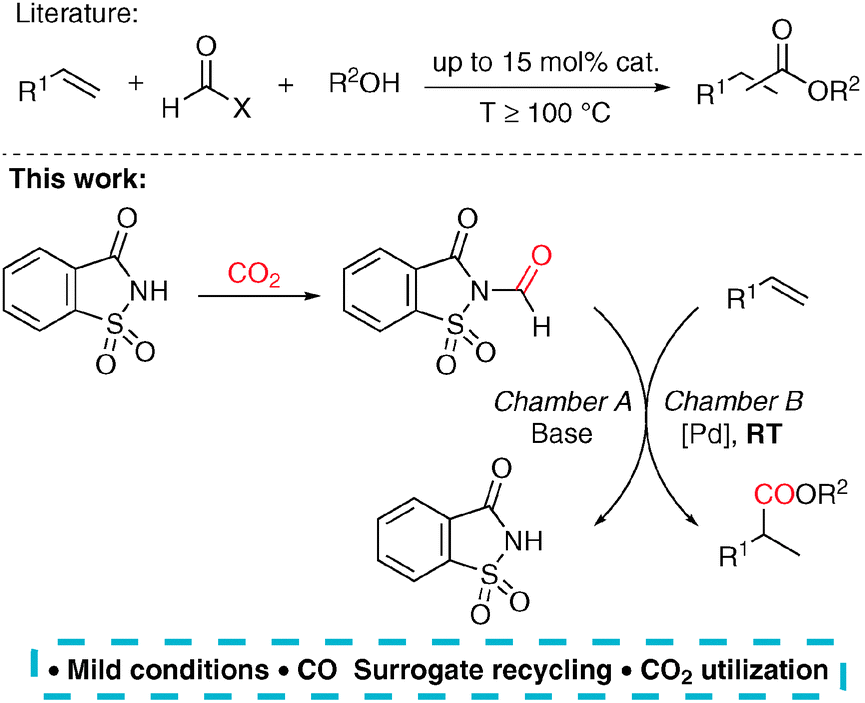 | ||
| Scheme 1 Use of CO surrogates in alkoxycarbonylations of alkenes. Our work: indirect utilization of CO2 as a C1 source under ambient reaction conditions using N-formylsaccharin as a CO transfer reagent. | ||
First, the setup for the ex situ generation of CO and simultaneous alkoxycarbonylation was investigated. The reactions were performed in two-chamber pressure tubes developed by Skrydstrup.10a Carbon monoxide (max. 2.5 bar) was liberated from N-formylsaccharin (1) by treatment with a base in DMF at room temperature as previously described by Manabe and co-workers.12b,c The type of the base is important by means of reproducibility and control of the decarbonylation. Therefore, the solid Na2CO3 was used due to a slower reaction than with triethylamine. The choice of the carbonylation catalyst is based on the industrial production of methyl propionate from ethylene, which utilizes a palladium precursor and ligand dtpbx (2).13 Thus, the initial experiments using styrene and methanol as substrates were performed in order to identify the ideal catalyst and acid suitable for the carbonylation under room temperature and low pressure (Table 1). First, several acidic co-catalysts were tested. Interestingly, the most commonly used acids in this reaction such as para-toluene- and methanesulfonic acid caused low reactivity and moderate selectivity (Table 1, entries 1 and 2). So far, we don’t know the reason for the difference between the conversion and the yield. The much weaker benzoic acid led to even lower yield and regioselectivity (Table 1, entry 3). Remarkably, use of acids with pKa value in DMSO of 3.5 resulted in improved activity and high preference for the branched product. The reaction with TFA provided the esters 5aa in moderate yield and high selectivity after 2 h reaction time (Table 1, entry 4). Higher yield and slightly lower selectivity were observed in the reaction with racemic BINOL-phosphoric acid (BNPA, 3), which was chosen for further studies (Table 1, entry 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Pd source | HX | pKa (DMSO) |
b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :lb :lb |
Conv.b [%] | Yieldb [%] |
|
a Reaction conditions: chamber A: CO generation (max 2.5 bar): 1 (2.13 mmol, 449 mg), Na2CO3 (3.19 mmol, 340 mg) in DMF (1 mL); chamber B: styrene (1.00 mmol, 115 μL, 1 M solution), 1:3 MeOH:DCM (v/v), 0.5 mol% [Pd], 2 mol% dtbpx (20 μmol, 7.9 mg), 7.5 mol% HX, RT, 2 h.
b Determined by quantitative GC-FID analysis of the crude reaction mixture.
|
||||||
| 1 | Pd(dba)2 | pTsOH | 7.1 | 71:29 |
27 | 9 |
| 2 | Pd(dba)2 | MsOH | 1.6 | 69:31 |
28 | 6 |
| 3 | Pd(dba)2 | PhCOOH | 11.1 | 51:49 |
21 | 2 |
| 4 | Pd(dba)2 | TFA | 3.5 | 95:5 |
64 | 38 |
| 5 | Pd(dba)2 | 3 | 3.4 | 88:12 |
57 | 56 |
| 6 | Pd(acac)2 | 3 | 3.4 | — | 14 | 0 |
| 7 | PdCl2 | 3 | 3.4 | — | 18 | 0 |
| 8 | Pd(PPh3)4 | 3 | 3.4 | 93:7 |
11 | 1 |
| 9 | Pd(OAc)2 | 3 | 3.4 | 92:8 |
16 | 2 |
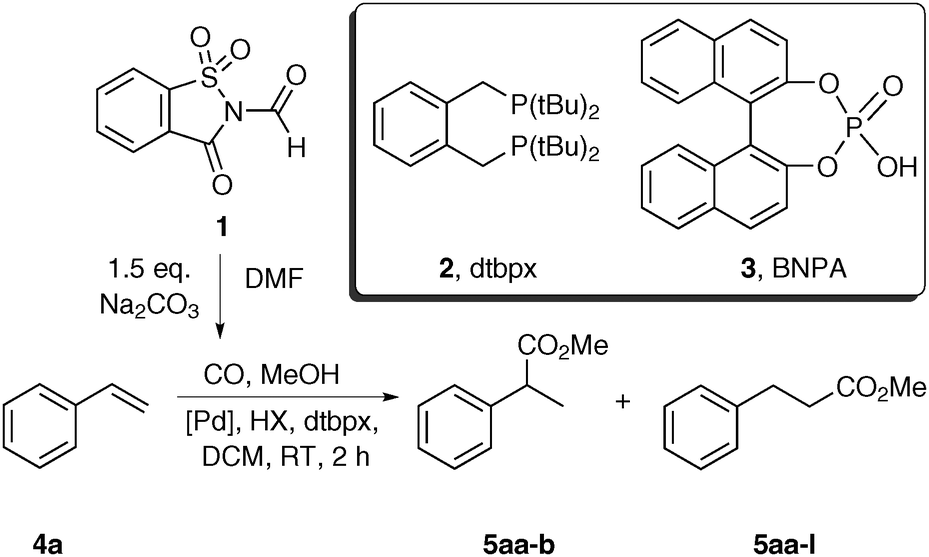
The examination of several palladium precursors revealed that best results were obtained using Pd(dba)2 (Table 1, entries 5–9). Thus, we were able to find mild reaction conditions for the selective production of the branched ester 5aa from styrene, which is in contrast to the literature-known carbonylations with the dtbpx ligand, which preferably provide the linear esters.5b,7e A notable exception is the work of Tanaka, however only one example is given.11e
Recently, concerns emerged that compared to other lower-molecular weight CO surrogates, 1 only contains 13% CO relative to its molecular weight.7e However, this problem can be alleviated by choice of an efficient recovery and regeneration process. The spent CO generation solution contains the sodium saccharinate (6), from which saccharin (7) can be precipitated in 87% yield by addition of HCl (Scheme 2). N-Formylsaccharin was synthesized by the formylation of 7 with the in situ-generated mixed anhydride 10 in the presence of 10 mol% of a base (pyridine or imidazole) in good yield with excellent chemoselectivity.14 This procedure can be used for a convenient synthesis of 1 at up to 20 g scale (92%).
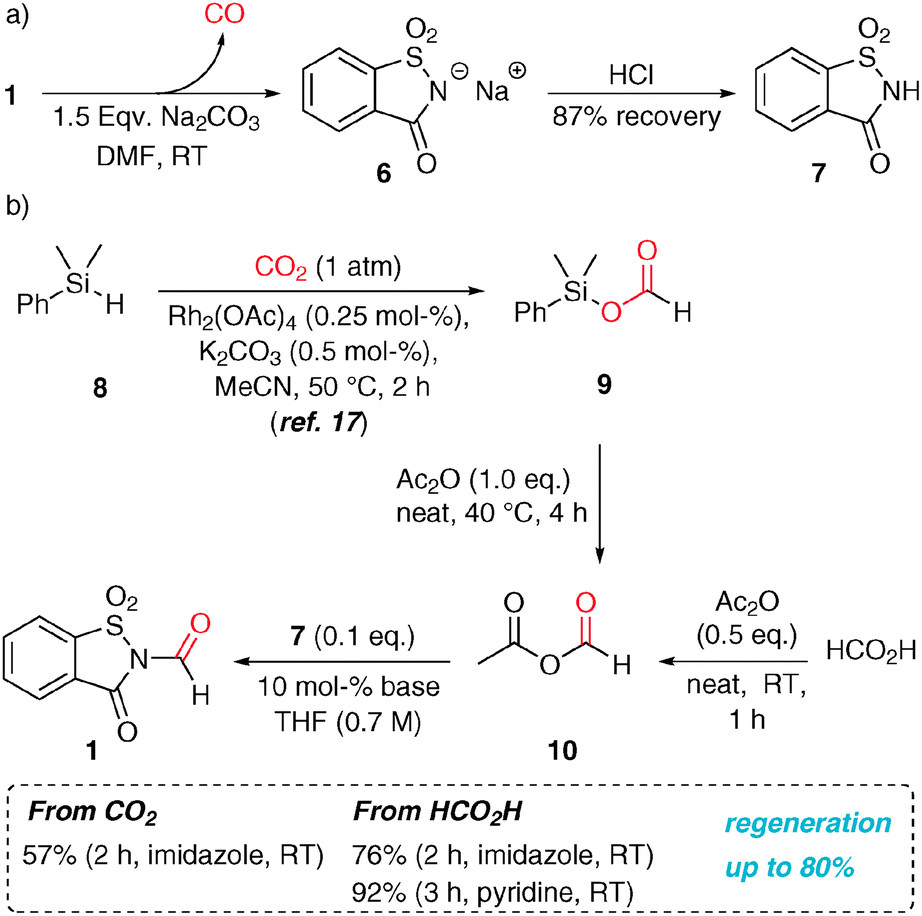 | ||
| Scheme 2 (a) Base-mediated decarbonylation of N-formylsaccharin. (b) Base-catalysed N-formylation of 7, where the formyl moiety either stems from formic acid or CO2. | ||
We were additionally intrigued by the possibility to synthesize the CO surrogate from carbon dioxide, since its utilization as a C1 source in the synthesis represents one of the main goals and challenges of modern organic chemistry.15 However, the transformations of CO2 usually require forcing reaction conditions. Therefore, we chose Rh-catalysed CO2 hydrosilylation under mild conditions as a basis for our efforts.16 Thus, silyl formate 9 was prepared following the literature procedure and it was subsequently converted to the anhydride 10 quantitatively at 40 °C. This mixture was then employed to synthesize 1 under the aforementioned conditions, albeit in somewhat lower yields possibly due to the presence of PhMe2SiOAc. In summary, this methodology allows for the capture of CO2 as a solid CO surrogate by a hydrosilylation/transacylation/formamidation sequence.
With reliable procedures for the recycling of N-formylsaccharin and carbonylation in hand, we proceeded to investigate the scope and limitations of the catalytic system. First, the influence of the alcohol component on the overall reactivity and regioselectivity in the alkoxycarbonylation of styrene was tested (Table 2, entries 1–7). Isolated yields decreased from primary (5aa, 5ab, 5af; Table 2 entries 1–3 and 7) to secondary alcohols (5ac, 5ae; Table 2 entries 4 and 6), while tBuOH provided no carbonylated product 5ad (Table 2, entry 5), which is in agreement with the accepted mechanism of the reaction, according to which the final alcoholysis of the Pd–acyl complex is the rate-determining step.17
|
|
||||
|---|---|---|---|---|
| Entry | Ar | R |
b:lb |
Yieldc [%] |
|
a Reaction conditions: Chamber A: CO generation (max 2.5 bar): 1 (2.13 mmol, 449 mg), Na2CO3 (3.19 mmol, 340 mg) in DMF (1 mL); chamber B: styrene (1.00 mmol, 115 μL, 1 M solution), 1:3 ROH:DCM (v/v), Pd(dba)2 (5.0 μmol, 2.9 mg), dtbpx (20 μmol, 7.9 mg), rac-BNPA (75 μmol, 26 mg), RT, 14 h.
b Determined by GC-FID analysis of the crude reaction mixture.
c Isolated yield.
d DCE as solvent.
e With Pd(dba)2 (10.0 μmol).
f The double ester was obtained.
g 50 °C, deacetylated product (ArOH).
h With 10.0 μmol Pd(dba)2.
i 5:95 MeOH:DCM (v/v), the lactone was obtained.
|
||||
| 1, 5aa | Ph | Me | 88:12 |
76 |
| 2, 5aad | Ph | Me | 87:13 |
80 |
| 3, 5ab | Ph | Et | 93:7 |
84 |
| 4, 5ac | Ph | iPr | 90:10 |
15 |
| 5, 5ad | Ph | tBu | — | 0 |
| 6, 5ae | Ph | Cy | 93:7 |
21 |
| 7, 5af | Ph | Bn | 82:18 |
79 |
| 8, 5ba | 3-Me-C6H4 | Me | 91:9 |
92 |
| 9, 5ca | 3-OMe-C6H4 | Me | 89:11 |
76 |
| 10, 5da | 4-Me-C6H4 | Me | 91:9 |
55 |
| 11, 5eae | 4-tBu-C6H4 | Me | 94:6 |
89 |
| 12, 5fa | 4-OMe-C6H4 | Me | 93:7 |
97 |
| 13, 5gaf | 4-COOH-C6H4 | Me | 77:23 |
21 |
| 14, 5ha | 4-NO2-C6H4 | Me | — | 0 |
| 15, 5iae | 4-Cl-C6H4 | Me | 92:8 |
90 |
| 16, 5jae | 4-OAc-C6H4 | Me | 93:7 |
43 |
| 17, 5kae,g | 4-OAc-C6H4 | Me | 86:14 |
57 |
| 18, 5la | 2-OMe-C6H4 | Me | 64:56 |
66 |
| 19, 5lae | 2-OMe-C6H4 | Me | 41:59 |
60 |
| 20, 5mah | 2-OAc-C6H4 | Me | 39:61 |
59 |
| 21, 5na | 2-Me-C6H4 | Me | 50:50 |
5 |
| 22, 5oa | 2-CF3-C6H4 | Me | — | 0 |
| 23, 5pi | 2-OH-C6H4 | Me | — | 16 |
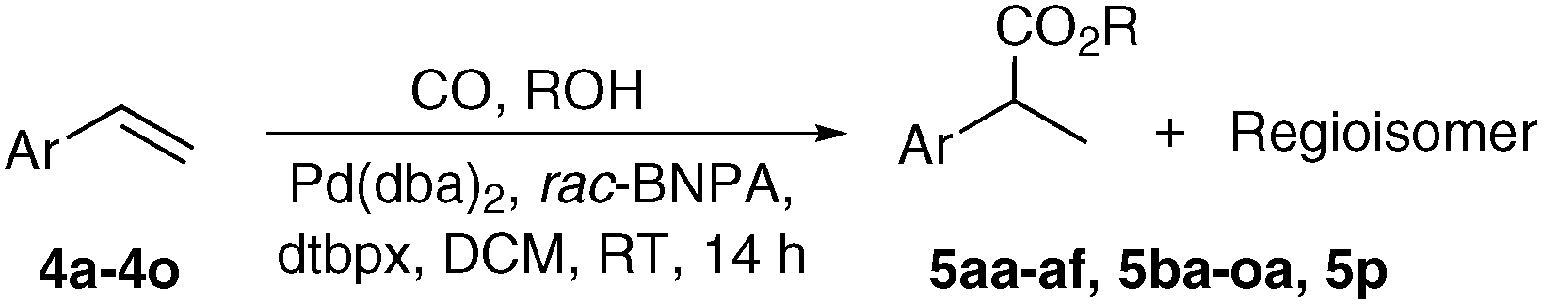
Next, the reactivity profile of various styrene derivatives in the methoxycarbonylation was examined (Table 2, entries 8–23). In general, meta- or para- alkyl and methoxy substituted styrene derivatives (Table 2, entries 8–12) gave high yields (up to 97%) with good to excellent branched-selectivity (89:11 to 94:6 b:l). The presence of an acidic function resulted in lower yield of 5ga and selectivity, probably due to a combination of electronic effect and acidity (Table 2, entry 13). Also, the carboxylic acid group was esterified with methanol. While an alkene with a strongly electron-withdrawing nitro group was not converted at all (Table 2, entry 14), a styrene with a moderately electron-withdrawing chloride in the para-position provided the product in 90% yield and 92% branched selectivity (Table 2, entry 15). Lower yields of the desired product 5ja and its deacetylated analogue 5ka (ArOH) were obtained from p-acetoxy-substituted styrene (Table 2, entries 16 and 17).
Due to their steric hindrance, ortho-substituted styrenes (Table 2, entries 18–21) gave only moderate yields and selectivities of the corresponding products. Furthermore, the combination of steric demand with electron-withdrawing properties led to no reactivity (Table 2, entry 22). It is noteworthy that 2-vinylphenol (4p) furnished the 5-membered cyclocarbonylated product 5p even in the presence of MeOH, albeit in low yield (Table 2, entry 23). In some cases, reactions giving low yields were improved either by doubling the catalyst loading or increasing the reaction temperature to 50 °C.
Finally, we investigated the reactivity of other olefin types in the carbonylation with methanol or benzyl alcohol (Table 3). As expected, the linear product was formed exclusively from methyl methacrylate (11), albeit in moderate yield. A similar result was obtained with another geminally disubstituted alkene 16. On the other hand, vinyl acetate (12) provided selectively the branched product, in contrast to similar catalyst systems using MsOH, where a maximal b:l ratio of 78:12 of the ester product was obtained.11e,18 In contrast, the transformation of N-vinylphthalimide (15) resulted in the formation of both regioisomers in 1:1 ratio. Interestingly, both β-methyl styrene (14) and allyl benzene (13) led to the same mixture of regioisomers. Only the benzylic and the terminal position were carbonylated in 1:1 ratio, which demonstrates the competition between the minimization of steric clashes and the formation of a stabilized η3-benzylpalladium species. Both terminal and internal aliphatic alkenes 17 and 18 were successfully transformed to the linear esters 27 (methyl) and 28 (benzyl), however higher catalyst loading and 50 °C was necessary to achieve complete conversion. The isomerization of the internal double bond and carbonylation of the terminal position was also observed in the case of oleic ester 19, however the isolated yield of the diester was low. Lastly, we also examined the methoxycarbonylation of ethinylbenzene (20), but disappointingly a polymerization was taking place, which resulted in a low isolated yield of the unsaturated ester.
|
a Reaction conditions: chamber A: CO generation (max 2.5 bar): 1 (2.13 mmol, 449 mg), Na2CO3 (3.19 mmol, 340 mg) in DMF (1 mL); chamber B: alkene (1.00 mmol, 1 M solution), 1:3 MeOH:DCM (v/v), 0.5 mol% Pd(dba)2 (5.0 μmol, 2.9 mg), 2 mol% dtbpx (20 μmol, 7.9 mg), 7.5 mol% rac-BNPA (75 μmol, 26 mg), RT, 14 h. Isolated yields are given; the bracketed values refer to regioselectivities (b:l) measured by GC-FID analysis of the crude reaction mixture.
b With BnOH:solvent (1:3).
c Regioisomer ratios refer to the benzylic and terminal ester, respectively.
d 0.75 mmol alkene.
e 50 °C, DCE as solvent.
|
|---|
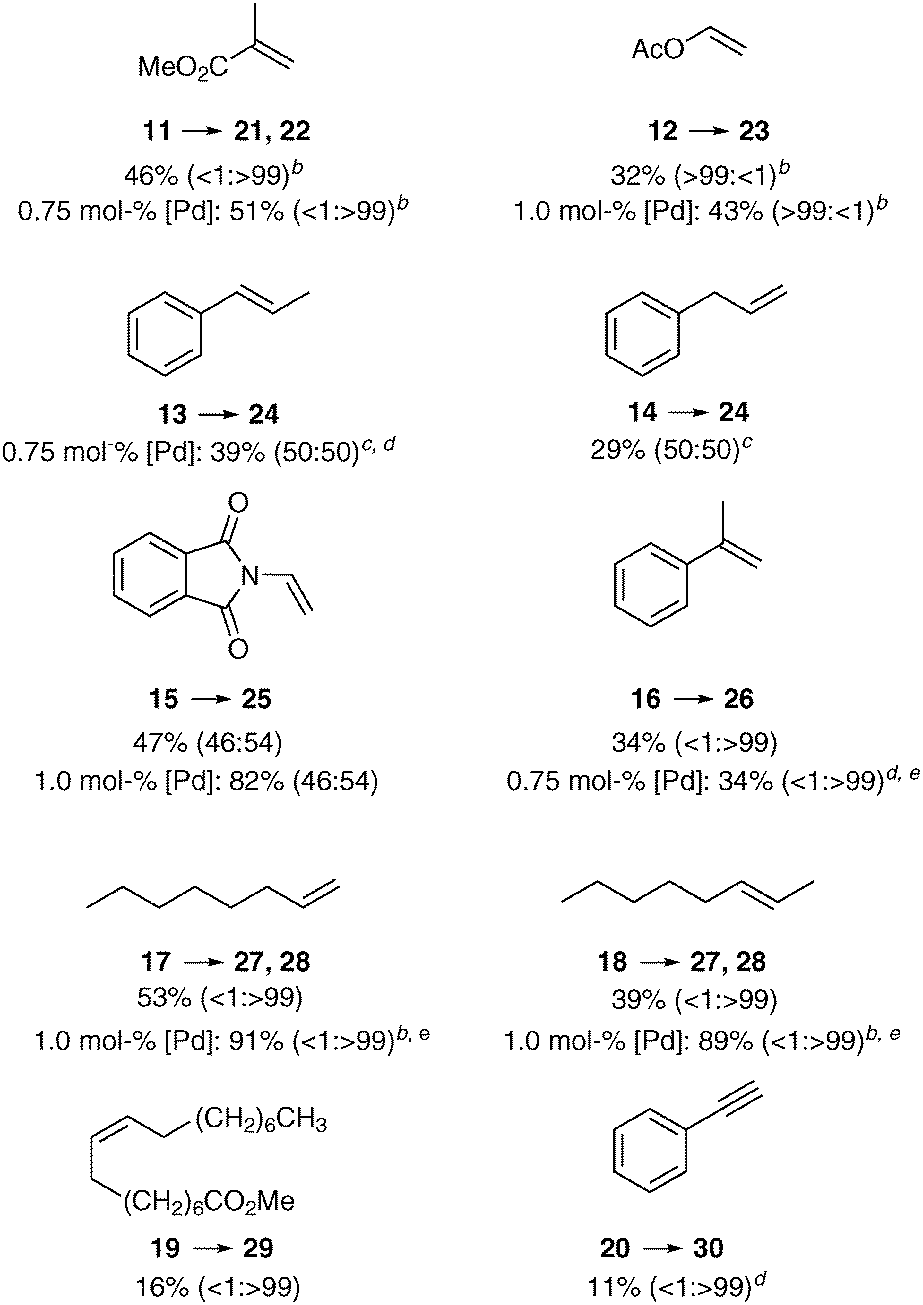
|
In conclusion, we have shown that N-formylsaccharin (1) is suitable for the ex situ generation of CO in Pd-catalysed hydroesterification reactions under mild reaction conditions. As opposed to an in situ CO generation approach, this ensures that the catalytic cycle of alkoxycarbonylations remains unchanged. The carbonylation catalyst is based on a Pd(0) precursor, bidentate phosphine ligand and a moderately strong acid, which enable a highly regioselective transformation of styrene derivatives to the corresponding branched esters. Moreover, also aliphatic and functionalized alkenes were successfully carbonylated. Notably, we have shown that the atom economy drawback associated with the use of 1 can also be addressed by the external CO generation method, since it allows for a straightforward recovery of saccharin and its conversion to 1 with acetic formic anhydride. Furthermore, it was demonstrated that CO2 can be transformed to a bench-stable CO surrogate in a single operation. With this proof-of-principle in hand, we are currently trying to develop a tandem procedure for the CO2 utilization.
We thank the Fonds der Chemischen Industrie (Liebig Fellowship I.F.) and the University of Regensburg for the support.
Notes and references
- (a) L. Kollár, Modern Carbonylation Methods, Wiley-VCH Verlag, Weinheim, 2008 Search PubMed; (b) C. F. J. Barnard, Organometallics, 2008, 27, 5402 CrossRef CAS; (c) X.-F. Wu and H. Neumann, ChemCatChem, 2012, 4, 447 CrossRef CAS PubMed; (d) G. Cavinato and L. Toniolo, Molecules, 2014, 19, 15116 CrossRef CAS PubMed; (e) W. Fang, H. Zhu, Q. Deng, S. Liu, X. Liu, Y. Shen and T. Tu, Synthesis, 2014, 1689 Search PubMed; (f) S. Quintero-Duque, K. M. Dyballa and I. Fleischer, Tetrahedron Lett., 2015, 56, 2634 CrossRef CAS PubMed.
- (a) M. Vasylyev and H. Alper, Synthesis, 2010, 2893 CAS; (b) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675 CrossRef CAS PubMed; (c) J. Pospech, I. Fleischer, R. Franke, S. Buchholz and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2852 CrossRef CAS PubMed; (d) M. Vilches-Herrera, L. Domke and A. Börner, ACS Catal., 2014, 4, 1706 CrossRef CAS.
- P. Kalck and M. Urrutigoïty, Inorg. Chim. Acta, 2015, 431, 110 CrossRef CAS PubMed.
- (a) T. Morimoto and K. Kakiuchi, Angew. Chem., Int. Ed., 2004, 43, 5580 CrossRef CAS PubMed; (b) H. Konishi and K. Manabe, Synlett, 2014, 1971 CAS; (c) L. Wu, Q. Liu, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 6310 CrossRef CAS PubMed.
- (a) H. Konishi, T. Ueda, T. Muto and K. Manabe, Org. Lett., 2012, 14, 4722 CrossRef CAS PubMed; (b) I. Fleischer, R. Jennerjahn, D. Cozzula, R. Jackstell, R. Franke and M. Beller, ChemSusChem, 2013, 6, 417 CrossRef CAS PubMed; (c) B. Li, S. Lee, K. Shin and S. Chang, Org. Lett., 2014, 16, 2010 CrossRef CAS PubMed; (d) Y. Wang, W. Ren, J. Li, H. Wang and Y. Shi, Org. Lett., 2014, 16, 5960 CrossRef CAS PubMed; (e) I. Profir, M. Beller and I. Fleischer, Org. Biomol. Chem., 2014, 12, 6972 RSC; (f) D.-S. Kim, W.-J. Park, C.-H. Lee and C.-H. Jun, J. Org. Chem., 2014, 79, 12191 CrossRef CAS PubMed.
- M. G. Mura, L. D. Luca, G. Giacomelli and A. Porcheddu, Adv. Synth. Catal., 2012, 354, 3180 CrossRef CAS PubMed.
- (a) G. Makado, T. Morimoto, Y. Sugimoto, K. Tsutsumi, N. Kagawa and K. Kakiuchi, Adv. Synth. Catal., 2010, 352, 299 CrossRef CAS PubMed; (b) E. Cini, E. Airiau, N. Girard, A. Mann, J. Salvadori and M. Taddei, Synlett, 2011, 199 CAS; (c) A. Kopfer, B. Sam, B. Breit and M. J. Krische, Chem. Sci., 2013, 4, 1876 RSC; (d) M. Uhlemann, S. Doerfelt and A. Börner, Tetrahedron Lett., 2013, 54, 2209 CrossRef CAS PubMed; (e) Q. Liu, K. Yuan, P.-B. Arockiam, R. Franke, H. Doucet, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 4493 CrossRef CAS PubMed.
- (a) E.-A. Jo, J.-H. Lee and C.-H. Jun, Chem. Commun., 2008, 5779 RSC; (b) J. J. Verendel, M. Nordlund and P. G. Andersson, ChemSusChem, 2013, 6, 426 CrossRef CAS PubMed; (c) S. H. Christensen, E. P. K. Olsen, J. Rosenbaum and R. Madsen, Org. Biomol. Chem., 2015, 13, 938 RSC.
- (a) K.-i. Tominaga, Catal. Today, 2006, 115, 70 CrossRef CAS PubMed; (b) T. G. Ostapowicz, M. Schmitz, M. Krystof, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 12119 CrossRef CAS PubMed; (c) K. Tsuchiya, J.-D. Huang and K.-i. Tominaga, ACS Catal., 2013, 3, 2865 CrossRef CAS; (d) L. Wu, Q. Liu, I. Fleischer, R. Jackstell and M. Beller, Nat. Commun., 2014, 5, 3091 Search PubMed; (e) Q. Liu, L. Wu, I. Fleischer, D. Selent, R. Franke, R. Jackstell and M. Beller, Chem. – Eur. J., 2014, 20, 6809 CrossRef CAS PubMed.
- (a) P. Hermange, A. T. Lindhardt, R. H. Taaning, K. Bjerglund, D. Lupp and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 6061 CrossRef CAS PubMed; (b) S. Korsager, D. U. Nielsen, R. H. Taaning and T. Skrydstrup, Angew. Chem., Int. Ed., 2013, 52, 9763 CrossRef CAS PubMed; (c) C. Lescot, D. U. Nielsen, I. S. Makarov, A. T. Lindhardt, K. Daasbjerg and T. Skrydstrup, J. Am. Chem. Soc., 2014, 136, 6142 CrossRef CAS PubMed; (d) Z. Lian, H. Yin, S. D. Friis and T. Skrydstrup, Chem. Commun., 2015, 51, 7831 RSC.
- (a) W. Clegg, G. R. Eastham, M. R. J. Elsegood, R. P. Tooze, X. L. Wang and K. Whiston, Chem. Commun., 1999, 1877 RSC; (b) G. R. Eastham, B. T. Heaton, J. A. Iggo, R. P. Tooze, R. Whyman and S. Zacchini, Chem. Commun., 2000, 609 RSC; (c) V. De La Fuente, M. Waugh, G. R. Eastham, J. A. Iggo, S. Castillón and C. Claver, Chem. – Eur. J., 2010, 16, 6919 CrossRef CAS PubMed; (d) P. Roesle, L. Caporaso, M. Schnitte, V. Goldbach, L. Cavallo and S. Mecking, J. Am. Chem. Soc., 2014, 136, 16871 CrossRef CAS PubMed; (e) H. Ooka, T. Inoue, S. Itsuno and M. Tanaka, Chem. Commun., 2005, 1173 RSC.
- (a) T. Cochet, V. Bellosta, A. Greiner, D. Roche and J. Cossy, Synlett, 2011, 1920 CAS; (b) T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2013, 15, 5370 CrossRef CAS PubMed; (c) T. Ueda, H. Konishi and K. Manabe, Angew. Chem., Int. Ed., 2013, 52, 8611 CrossRef CAS PubMed.
- M. W. G. R. Eastham, P. Pringle and T. P. W. Turner, WO2011083305, 2011.
- H. Yazawa and S. Goto, Tetrahedron Lett., 1985, 26, 3703 CrossRef CAS.
- (a) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS PubMed; (b) M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620 CrossRef CAS PubMed; (c) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- S. Itagaki, K. Yamaguchi and N. Mizuno, J. Mol. Catal. A: Chem., 2013, 366, 347 CrossRef CAS PubMed.
- G. Kiss, Chem. Rev., 2001, 101, 3435 CrossRef CAS PubMed.
- A. J. Rucklidge, G. E. Morris and D. J. Cole-Hamilton, Chem. Commun., 2005, 1176 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc05012j |
| This journal is © The Royal Society of Chemistry 2015 |