
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coverage-driven dissociation of azobenzene on Cu(111): a route towards defined surface functionalization†
Martin
Willenbockel
ab,
Reinhard J.
Maurer‡
 c,
Christopher
Bronner§
de,
Michael
Schulze
de,
Benjamin
Stadtmüller¶
ab,
Serguei
Soubatch
*ab,
Petra
Tegeder
d,
Karsten
Reuter
c and
F.
Stefan Tautz
ab
c,
Christopher
Bronner§
de,
Michael
Schulze
de,
Benjamin
Stadtmüller¶
ab,
Serguei
Soubatch
*ab,
Petra
Tegeder
d,
Karsten
Reuter
c and
F.
Stefan Tautz
ab
aPeter Grünberg Institut (PGI-3), Forschungszentrum Jülich, 52425 Jülich, Germany. E-mail: s.subach@fz-juelich.de
bJülich Aachen Research Alliance (JARA), Fundamentals of Future Information Technology, 52425 Jülich, Germany
cChair of Theoretical Chemistry and Catalysis Research Center, Technische Universität München, Lichtenbergstrasse 4, 85747 Garching, Germany
dPhysikalisch-Chemisches Institut, Ruprecht-Karls-Universität Heidelberg, Im Neuenheimer Feld 253, 69120 Heidelberg, Germany
eFreie Universität Berlin, Fachbereich Physik, Arnimallee 14, 14195 Berlin, Germany
First published on 28th August 2015
Abstract
We investigate the surface-catalyzed dissociation of the archetypal molecular switch azobenzene on the Cu(111) surface. Based on X-ray photoelectron spectroscopy, normal incidence X-ray standing waves and density functional theory calculations a detailed picture of the coverage-induced formation of phenyl nitrene from azobenzene is presented. Furthermore, a comparison to the azobenzene/Ag(111) interface provides insight into the driving force behind the dissociation on Cu(111). The quantitative decay of azobenzene paves the way for the creation of a defect free, covalently bonded monolayer. Our work suggests a route of surface functionalization via suitable azobenzene derivatives and the on surface synthesis concept, allowing for the creation of complex immobilized molecular systems.
There is considerable interest in developing molecular assemblies on surfaces for a multitude of applications such as photovoltaics, biomaterials, sensors, biochips, and molecular electronics, but also to control surface reactions and tailor surface properties.1,2 An established route to modify surfaces is to use self-assembled monolayers (SAMs). SAMs are molecular assemblies that are formed spontaneously via the adsorption of a surfactant with a specific affinity of its anchoring group to a substrate.1,2 However, the defect density and the degree of order in SAMs strongly depend on the surface roughness as well as on the molecular components involved, i.e., the anchor-/linker-system and the terminal end group (functional group).
In this communication, employing a combination of X-ray photoelectron spectroscopy (XPS), normal incidence X-ray standing waves (NIXSW) and density functional theory (DFT), we demonstrate a coverage-driven quantitative dissociation of azobenzene on Cu(111), leading to the formation of a well-defined covalently bonded and ordered phenyl nitrene monolayer, which can serve as a basis for various surface functionalizations. Based on indirect evidence, dissociation was recently also proposed for an azobenzene derivative on Cu(100).3 Moreover, azobenzene dissociation is not limited to Cu surfaces, but also expected to occur on a wider range of substrates, e.g. TiO2.4–6
Contrary, we observed no decay of azobenzene on the Ag(111) surface. The essential difference between Ag(111) and Cu(111) turns out to be the balance between molecule–molecule and molecule–substrate interactions: in the copper case, the increased molecule–molecule interaction in denser structures leads to dissociation, because the stronger N–Cu bond leaves no structural alternative, while in the silver case the same interaction pushes the structure of the molecule toward a deformed trans configuration.
A common approach to investigate the adsorption behaviour of organic molecules on surfaces is XPS. Based on chemically resolved core level binding energies, it allows for insights into the chemical properties of any given adsorbate. An example is shown in Fig. 1, where the N1s core level spectra of an azobenzene submonolayer, monolayer and multilayer are displayed. From the observation of several distinct peaks, and the pronounced chemical shifts between them, it is immediately clear that azobenzene exists in several chemically different configurations and/or environments on Cu(111). In particular, the occurrence of a second peak in the monolayer spectrum, shifted by 1.5 eV against the submonolayer peak, suggests the existence of a reacted species in the monolayer.
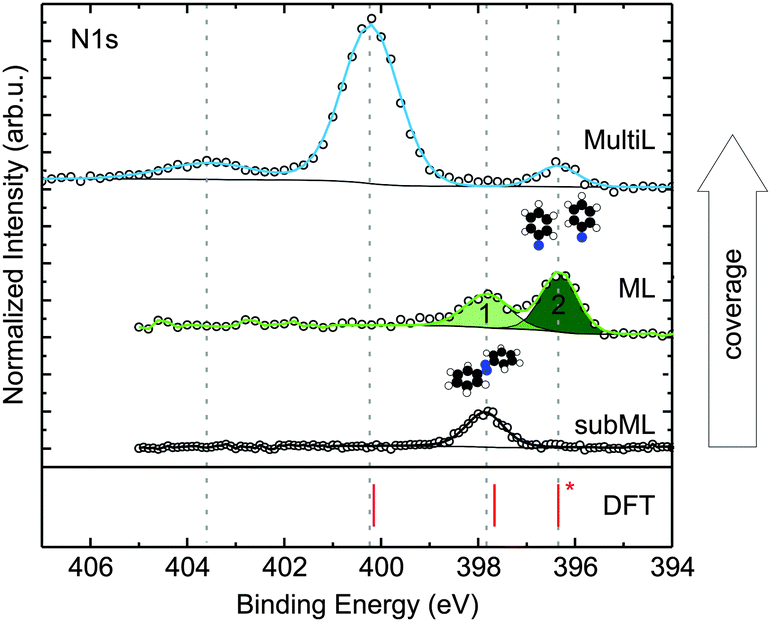 | ||
| Fig. 1 N1s XPS spectra for submonolayer, monolayer, and multilayer of azobenzene on Cu(111). Coverages were determined by XPS. Spectra were normalized to the background at low binding energy and shifted on the vertical axis for clarity. 1 and 2 refer to the peaks for which the results of a NIXSW analysis are displayed in Fig. 2. In the bottom panel calculated results for the N1s binding energies are shown. The calculated binding energies correspond to free (gas-phase) azobenzene (400.1 eV), azobenzene adsorbed on Cu(111) (397.6 eV), and phenyl nitrene on Cu(111) (396.3 eV), the latter two are corrected for the surface work function (see also ESI†). | ||
XPS spectra are certainly instructive, but without additional information, e.g. on the geometry of the adsorbate, it is nevertheless impossible to reach definite conclusions regarding the nature of the various species. This shortcoming of XPS can be overcome by NIXSW technique.7,8 NIXSW employs the interference of an X-ray beam with its Bragg-reflected wave to form a standing wave field above the surface of a crystalline sample. This spatially modulated X-ray intensity allows for a chemically sensitive, precise measurement of vertical adsorption distances. Each vertical adsorption height leads to a characteristic profile of the XPS photoelectron yield as a function of the X-ray energy around the Bragg condition. Examples of such profiles for nitrogen in azobenzene are shown in Fig. 2. Since the technique is based on core level spectra, it has the same chemical resolution as XPS,7,8i.e. each chemically resolved species in XPS can be assigned a definite adsorption height.
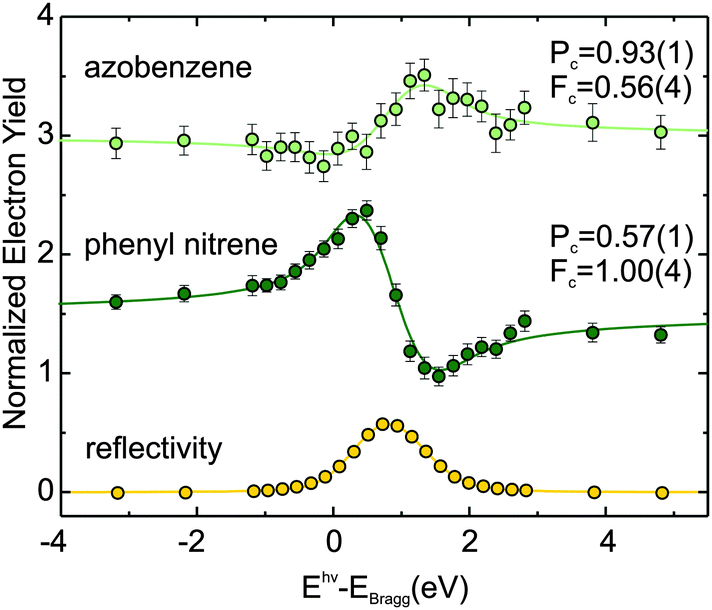 | ||
| Fig. 2 NIXSW N1s electron yield curves (as a function of photon energy Ehν relative to the Cu(111) Bragg energy EBragg) for the monolayer coverage regime (green curve in Fig. 1) with intact azobenzene (1) and phenyl nitrene (2) coexisting on the Cu(111) surface. The fits have been carried out with the program Torricelli.12,13 The X-ray reflectivity curve is shown in yellow. Coherent positions Pc and fractions Fc derived from the fit are also displayed. From the coherent positions, the corresponding adsorption heights can be determined.7,8 The distinct profiles of the electron yield curves reveal a change of the nitrogen adsorption height upon dissociation and allow for the establishment of an unambiguous link between the surface species and the corresponding core level binding energy in the XPS spectrum in Fig. 1. | ||
For azobenzene on Cu(111), considering nitrogen first, the following picture emerges: a submonolayer of azobenzene on Cu(111) gives rise to a single N1s peak at 397.8 eV, shown in Fig. 1 (black). Compared to the bulk signal of azobenzene,9 which corresponds to the N1s peak at 400.2 eV in the multilayer spectrum (blue), the submonolayer species exhibits a shift of 2.4 eV toward lower binding energy; the direction of the shift is consistent with screening by the metal surface, while its strength suggests a chemical interaction between nitrogen and the copper surface. Indeed, in our NIXSW experiments we find the diazo bridge 2.02(2) Å above the topmost Cu(111) Bragg plane. This vertical adsorption distance constrains the possible N–Cu interatomic distances to within the range from 2.02(5) Å to 2.50(5) Å, depending on the adsorption site and a possible Cu relaxation.10 Since these bond lengths are decidedly smaller than the sum of the copper and nitrogen van der Waals radii (3.0 Å),11 we can conclude that a chemical bond forms between the diazo bridge and the Cu(111) surface.
If the coverage is increased to the monolayer range, a new N1s peak appears in the XPS spectrum at 396.3 eV (green), in coexistence with the peak at 397.8 eV. The further shift by 1.5 eV towards lower binding energy reflects an increased interaction between nitrogen and the Cu(111) surface. In our NIXSW analysis (Fig. 2), a drastic change of the adsorption geometry is indeed immediately obvious from the different electron yield profiles (light and dark green). A fit reveals a vertical nitrogen distance of only 1.17(4) Å above the topmost Cu(111) Bragg plane. Only a threefold hollow adsorption site is consistent with such a small adsorption height; hence, the N–Cu bond length of the new species must be 1.89(7) Å. Interestingly, this bond length is similar to the one predicted for the dissociative adsorption of N2 on Cu(111).17 Together with a mean carbon height of 4.25(4) Å, extracted from the C1s NIXSW yield profiles (see Table 1), this finding indicates that azobenzene must have dissociated to form upright standing phenyl nitrene.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14,15). Three heights are given, namely the height of the nitrogen atoms (dCu−N), the height of the phenyl ring carbon atoms bound to the nitrogen atoms (dCu−CN), and the mean height of remaining phenyl ring carbon atoms (bound to carbon atoms) (dCu−CC). All heights are given relative to the topmost Cu(111) Bragg plane. Two angles are given, namely the dihedral tilt and twist angles ω (CNNC) and β (NNCC), as indicated in Fig. 3. The experimental angles have been determined by Fourier vector analysis following ref. 16
14,15). Three heights are given, namely the height of the nitrogen atoms (dCu−N), the height of the phenyl ring carbon atoms bound to the nitrogen atoms (dCu−CN), and the mean height of remaining phenyl ring carbon atoms (bound to carbon atoms) (dCu−CC). All heights are given relative to the topmost Cu(111) Bragg plane. Two angles are given, namely the dihedral tilt and twist angles ω (CNNC) and β (NNCC), as indicated in Fig. 3. The experimental angles have been determined by Fourier vector analysis following ref. 16
| d Cu−N (Å) | d Cu−CN (Å) | d Cu−CC (Å) | ω (deg) | β (deg) | ||
|---|---|---|---|---|---|---|
| Azobenzene | XSW Experiment | 2.02(2) | 2.23(6) | 2.36(2) | 18.8 | 3.8 |
| DFT+vdWsurf | 2.03 | 2.15 | 2.28 | 11.0 | 3.3 | |
| Phenyl nitrene | XSW Experiment | 1.17(4) | — | 4.25(4) | ||
| DFT+vdWsurf | 1.18 | 2.58 | 4.27 | |||
We have calculated the adsorption structure of azobenzene and the dissociation product phenyl nitrene on Cu(111) with dispersion-corrected DFT (cf. ESI† for details), employing the DFT+vdWsurf method,14,15,18,19 known for its reliable description of energetics and geometry.15,20,21 The results, displayed in Table 1, show an excellent agreement between the NIXSW experiment and theory for all key structural parameters. DFT thus confirms the coexistence of two species, one intact azobenzene, the other dissociated phenyl nitrene, in the monolayer.
To complete the argument, we have used the calculated structures to simulate N1s binding energies and compare them to the experimentally observed ones (cf. ESI† for details). Indeed, we are able to accurately predict the relative binding energy shifts between the species in Fig. 1. Due to the approximate semi-local description of exchange and correlation, the absolute binding energies are systematically underestimated, for which we correct by rigidly shifting our calculated XPS binding energies, aligning the calculated phenyl nitrene peak with the lowest binding energy peak found in the experiment (396.3 eV). We find that the N1s binding energy calculated for the gas-phase molecule matches with the main peak at 400.1 eV in the multilayer spectrum. Hence, we can identify this peak as the N1s binding energy of azobenzene molecules in the multilayer, while the small feature at 403.6 eV most likely corresponds to a shake-up peak due to higher-order deexcitation processes.
Summarising up to this point, we have obtained from XPS, NIXSW and DFT a fully consistent picture of a coverage-induced dissociation of adsorbed azobenzene (Fig. 3a). The lateral structure of the system, investigated with low electron energy diffraction (LEED), is also consistent with this conclusion. No lateral order is found in the initial stages of adsorption. When the coverage is increased, an ordered phase with a point-on-line registry is found; its superstructure matrix is 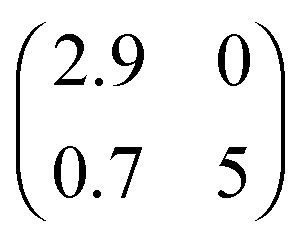 . We associate this phase with intact azobenzene molecules. Further increasing the coverage, we observe, after a transition range, commensurate order with a (4 × 4) superstructure, which also persists for the multilayer. Because this order is still observed after thermally desorbing the multilayer, the (4 × 4) structure can be assigned to the dissociated phenyl nitrene contact layer that remains on the surface. The presence of the phenyl nitrene contact layer below the multilayer is also evident in the multilayer XPS spectrum in Fig. 1. It should be noted that in the latter spectrum the peak of the intact azobenzene is absent. This indicates that the contact layer consists exclusively of dissociated molecules, i.e. the coverage-driven dissociation of azobenzene is quantitative.
. We associate this phase with intact azobenzene molecules. Further increasing the coverage, we observe, after a transition range, commensurate order with a (4 × 4) superstructure, which also persists for the multilayer. Because this order is still observed after thermally desorbing the multilayer, the (4 × 4) structure can be assigned to the dissociated phenyl nitrene contact layer that remains on the surface. The presence of the phenyl nitrene contact layer below the multilayer is also evident in the multilayer XPS spectrum in Fig. 1. It should be noted that in the latter spectrum the peak of the intact azobenzene is absent. This indicates that the contact layer consists exclusively of dissociated molecules, i.e. the coverage-driven dissociation of azobenzene is quantitative.
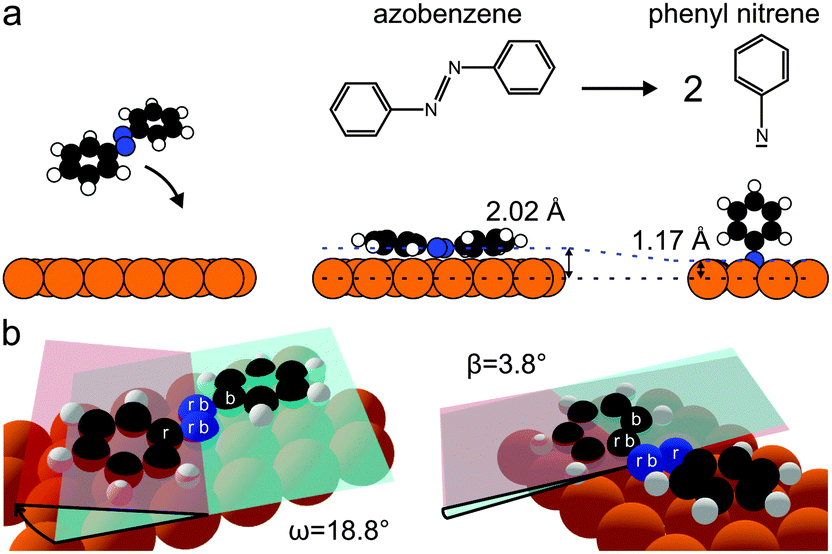 | ||
| Fig. 3 (a) Dissociation of azobenzene to phenyl nitrene, using structures from Table 1. Cu surface atoms are shown in orange, carbons in black, nitrogens in blue and hydrogens in white. (b) 3D structural model of azobenzene in the submonolayer adsorption geometry. The dihedral tilt and twist angles ω (CNNC) and β (NNCC) are visualized by the crossing of the red and blue semitransparent planes. The atoms which span the red and blue planes are indicated by an r and b. | ||
We now turn to the crucial question: what drives the dissociation of azobenzene upon adsorption, and what is the difference between the adsorption of azobenzene on Cu(111), where dissociation occurs, and Ag(111), where the molecule stays intact? To address this issue, we make use of a recent development in the analysis of NIXSW data. In their Fourier vector analysis Mercurio et al.16 go beyond the established calculation of adsorption heights8 and compare both the coherence and the phase of NIXSW data to NIXSW simulations of different molecular geometries. In this way, detailed adsorption geometries of large and complex molecules can be retrieved, even if the corresponding species cannot be resolved by their chemical shifts in XPS. Evidently, this approach is particularly attractive for organic molecules such as azobenzene which contain many carbon atoms that cannot be distinguished in XPS, due to the smallness or even absence of chemical shifts.
Applying the Fourier vector analysis to the carbon atoms in azobenzene, the following picture emerges (Fig. 3): upon adsorption on Cu(111) the flat gas-phase trans-azobenzene undergoes a conformational distortion; the phenyl ring tilts by ω = 18.8° and twists by β = 3.8°. Here ω and β are the dihedral angles defined in Fig. 3b. We note that theory predicts a very similar distortion (Table 1). The fact that the distortion mainly occurs in ω is a consequence of the low adsorption height of the diazo bridge, which pulls the phenyl rings into the repulsive regime until they respond by tilting out of the surface plane.
On Ag(111), where azobenzene also distorts, two trends can be discerned when the coverage is increased: first, the distortion of the molecule increases with rising coverage and second, the diazo bridge is lifted from the surface.22 The driving force behind the distortion is the increasing adsorption energy per surface area that can be gained when the footprint of the molecule on the surface is reduced and thus the molecular packing density is increased; this overcompensates the loss of adsorption energy per molecule in the distorted configuration.22 Accordingly, on Ag(111) the largest distortion occurs in β, i.e. the twisting angle that is most effective in reducing the molecular footprint.23
It is reasonable to expect that a similar trade-off between adsorption energies per area and per molecule is at work for azobenzene on Cu(111) as well. However, there is a crucial difference: on Cu(111) the diazo bridge of the isolated molecule adsorbs at a much lower distance to the surface, even if we correct the values for the difference in the van-der-Waals radii of Cu (1.4 Å11) and Ag (1.72 Å11), and moreover, azobenzene is already more strongly bent in the low coverage regime, especially in ω. Following the idea of the trade-off mentioned above, an increased coverage should lead to an enhanced distortion in β. However, an increase in β will always come at the cost of increasing the adsorption height of the diazo bridge. But this is not possible on Cu(111), simply because this bond is much stronger than on Ag(111),24,25 too strong in fact to yield to an increased twisting. The system therefore has to make a choice: either the diazo bridge and its adsorption height remain unchanged, but then the molecular footprint cannot be reduced, or the molecule has to find a different way to reduce its footprint, but this requires a modification of the N–Cu bond. Apparently, the latter option is energetically preferred on Cu(111): the diazo bridge dissociates, each nitrogen atom moves to a threefold hollow site and the phenyl rings flip into the vertical.
In conclusion, Fig. 4 illustrates the behaviour of azobenzene on Ag(111) and Cu(111) as it emerges from our analysis, both with regard to individual molecules and the entire layer: for azobenzene/Ag(111), the relative ‘elasticity’ of the N–Ag bond allows for an increasing layer density by a continuously increasing molecular distortion (upper left). The increasing average adsorption height of the layer therefore corresponds one-to-one to an increasing individual adsorption height of intact azobenzene molecules. For azobenzene/Cu(111), on the other hand, the ‘rigidity’ of the stronger N–Cu bonds, both of the diazo bridge and the nitrene, precludes a continuously evolving molecular distortion and consequently adsorption height (bottom right); a discontinuous transition, effected by the dissociation, reduces the footprint of individual molecules (and also leads to a lower adsorption height of the nitrogen). This discontinuity implies that the increased layer density can only be realized by dissociating more and more azobenzene molecules. Hence, steric hindrance in increasingly dense layers drives the dissociation, and the observed continuous reduction of the nitrogen adsorption height (bottom left) follows simply from replacing a rising number of azobenzenes by phenyl nitrenes.
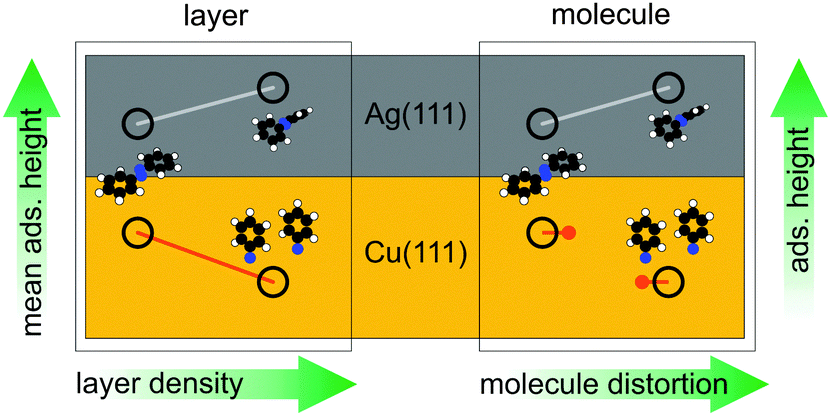 | ||
| Fig. 4 The behaviour of azobenzene on Ag(111) and Cu(111), both with regard to individual molecules (right) and the entire layer (left). | ||
The coverage-driven dissociation of azobenzene on Cu(111) is quantitative, i.e., the amount of non-transformed azobenzene molecules is below the detection limit of the surface sensitive XPS, thus allowing for a controlled functionalization of the copper surface. This unique formation of a defect-free monolayer opens a way for further functionalization via utilization of appropriate azobenzene derivatives, e.g. substituted at the phenyl rings in para position. The substituent may then serve as an active group in an on surface synthesis, facilitating the generation of complex molecular systems. This concept is also promising for the functionalization of other surfaces, e.g. TiO2 and may even be applicable under wet chemical conditions.
Experiments were carried out at the European Synchrotron Radiation Facility. We thank J. Duvernay, B. Detlefs, J. Zegenhagen, S. Stremlau, and E. Varene for their assistance and experimental support. Funding by the DFG through the SFB 658 is gratefully acknowledged. DFT calculations were carried out at the Leibnitz-Rechenzentrum under grant pr63ya.
References
- F. Schreiber, J. Phys.: Condens. Matter, 2004, 16, R881 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- M. Piantek, J. Miguel, A. Krüger, C. Navio, M. Bernien, D. K. Ball, K. Hermann and W. Kuch, J. Phys. Chem. C, 2009, 113, 20307–20315 CAS.
- S.-C. Li and U. Diebold, J. Am. Chem. Soc., 2010, 132, 64–66 CrossRef CAS PubMed.
- S.-C. Li, Y. Losovyj, V. K. Paliwal and U. Diebold, J. Phys. Chem. C, 2011, 115, 10173–10179 CAS.
- D. Kreikemeyer-Lorenzo, W. Unterberger, D. Duncan, T. Lerotholi and D. Woodruff, Surf. Sci., 2013, 613, 40–47 CrossRef CAS PubMed.
- J. Zegenhagen, Surf. Sci. Rep., 1993, 18, 202–271 CrossRef.
- D. P. Woodruff, Rep. Prog. Phys., 2005, 68, 743 CrossRef CAS.
- B. J. Lindberg and J. Hedman, Chem. Scr., 1975, 7, 155–166 CAS.
- S. A. Lindgren, L. Walldén, J. Rundgren and P. Westrin, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 29, 576–588 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- G. Mercurio, Study of Molecule-Metal Interfaces by Means of the Normal Incidence X-ray Standing Wave Technique, PhD thesis, Schriften des Forschungszentrums Jülich, Reihe Schlüsseltechnologien, vol. 49, 2012, available at www.fz-juelich.de/zb/juwel Search PubMed.
- Torricelli is an XSW data analysis and simulation program written by G. Mercurio; copies can be obtained from E-mail: s.tautz@fz-juelich.de.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef.
- V. G. Ruiz, W. Liu, E. Zojer, M. Scheffler and A. Tkatchenko, Phys. Rev. Lett., 2012, 108, 146103 CrossRef.
- G. Mercurio, R. J. Maurer, S. Hagen, F. Leyssner, J. Meyer, P. Tegeder, S. Soubatch, K. Reuter and F. S. Tautz, Front. Phys., 2014, 2, 13 Search PubMed.
- A. Soon, L. Wong, M. Lee, M. Todorova, B. Delley and C. Stampfl, Surf. Sci., 2007, 601, 4775–4785 CrossRef CAS PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CrossRef CAS.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS PubMed.
- G. Mercurio, O. Bauer, M. Willenbockel, N. Fairley, W. Reckien, C. H. Schmitz, B. Fiedler, S. Soubatch, T. Bredow, M. Sokolowski and F. S. Tautz, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 045421 CrossRef.
- W. Liu, F. Maaß, M. Willenbockel, C. Bronner, M. Schulze, S. Soubatch, F. S. Tautz, P. Tegeder and A. Tkatchenko, Phys. Rev. Lett., 2015, 115, 036104 CrossRef.
- G. Mercurio, R. J. Maurer, W. Liu, S. Hagen, F. Leyssner, P. Tegeder, J. Meyer, A. Tkatchenko, S. Soubatch, K. Reuter and F. S. Tautz, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 035421 CrossRef.
- We note that also for Ag(111) there is a good correspondence between experiment and theory. However, in contrast to the present case of azobenzene/Cu(111), where ground state calculations provide an excellent agreement with experimental structures, the calculation for azobenzene on Ag(111) requires thermal expansion taken into account, due to the weaker bond of the molecule to the surface22.
- E. McNellis, J. Meyer, A. D. Baghi and K. Reuter, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 035414 CrossRef.
- E. R. McNellis, J. Meyer and K. Reuter, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205414 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc05003k/ |
| ‡ Present address: Yale University, USA. |
| § Present address: University of California at Berkeley, USA. |
| ¶ Present address: Universität Kaiserslautern, Germany. |
| This journal is © The Royal Society of Chemistry 2015 |