
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A mild carbon–boron bond formation from diaryliodonium salts†
N.
Miralles
a,
R. M.
Romero
b,
E.
Fernández
*a and
K.
Muñiz
*bc
aDepartment Química Física i Inorgánica, University Rovira i Virgili, C/Marcellí Domingo s/n, 43007 Tarragona, Spain. E-mail: Mariaelena.fernandez@urv.cat
bInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: kmuniz@ICIQ.ES
cCatalan Institution for Research and Advanced Studies (ICREA), Pg. Lluís Companys 23, 08010 Barcelona, Spain
First published on 29th July 2015
Abstract
The direct metal-free borylation of diaryliodonium salts with diboron reagents is now demonstrated to be a feasible process toward formation of aryl boronic esters without any additive or catalysts, and it can be extended to a two-step C–C coupling of both aryl groups of the initial diaryliodonium reagent.
Due to their ready availability and high stability, diaryliodonium salts 1 constitute an attractive class of compounds that have been recognized as particularly versatile aryl transfer reagents.1 Consequently, they have been involved in a series of arylation reactions, which have been mostly aided by the presence of transition metal catalysts. While initially C–C bond forming reactions had been explored to a larger extent, carbon–heteroatom bond forming events based on diaryliodonium salts have been investigated in greater detail in recent years (Scheme 1, eqn (1)).2–10 Among all these accomplishments, carbon–boron bond formation toward aryl boronic acid derivatives is notably absent.
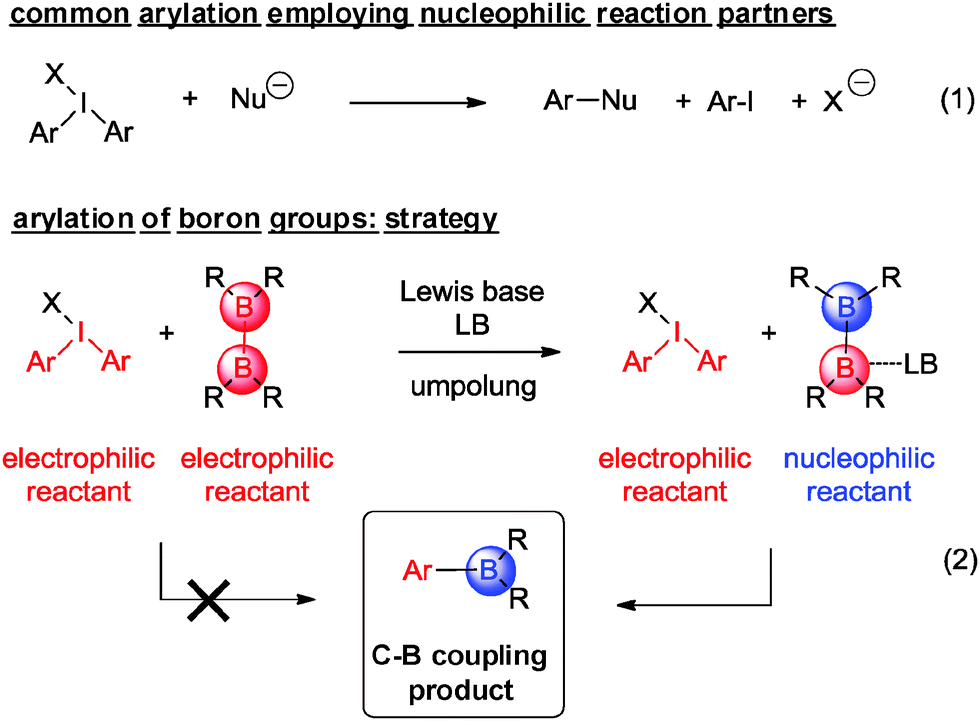 | ||
| Scheme 1 Conceptual approaches to carbon–heteroatom bond formation using diaryliodonium salts. | ||
Arylboronic acids and esters are key components for modern cross-coupling reactions and advanced synthetic transformations.11 Their high versatility has made C–C coupling events based on them one of the most versatile themes in the field,12 which was recognized by the Nobel prize for Suzuki in 2010.13 Common approaches to this type of reagents employ the original Miyaura-type borylation reaction between aryl halides and bis(pinacolato)diboron (pinB–Bpin, 2a) in the presence of catalytic amounts of palladium complexes. The key intermediate [LPd(Ar)(OAc)] undergoes a transmetallation process with 2a.14 Other transition metal complexes have catalyzed the aryl halide borylation15 complementing the C–H activation of arenes by borane reagents.16 We here report the unprecedented direct formation of aryl boronic esters, in particular aryl pinacolboranes, through an effective coupling between diaryliodonium salts and bis(pinacolato)diboron 2a under mild conditions and without any requirement for a metal promoter. In principle, the synthetic approach to such a type of direct C–B bond formation appears unconventional at first sight, as it involves the combination of two reactants that are of exceedingly electrophilic nature (Scheme 1, eqn (2)).
To accomplish the targeted carbon–boron bond formation, we envisioned that a Lewis base could be employed for the activation of the diboron reactant. Such an interaction should be favorable as it provides an umpolung of the native electrophilic boron atom. The resulting nucleophilic character17 at one of the boron centers then ensures for effective C–B bond formation with the electrophilic iodine(III) reagent. This approach is reminiscent of previous alkoxide base mediated reactions developed by some of us18,19 and others.20,21 A subsequent screening between various diphenyliodonium salts 1a–d and bis(pinacolato)diboron (2a) was undertaken and for the acetate derivative 1d confirmed the correctness of the initial hypothesis (Scheme 2). In agreement with previous experience,18,19 methanol was identified as the best solvent.22 For the reaction between acetate derivative 1d and 2a, the expected product 3a was formed as an isolated high yield of 77%. This successful transformation leads to two important conclusions: first, methanol as the solvent may play a crucial role in activating the diboron reagent through a state such as A. Secondly, the pronounced dependence of the reaction on the counterion of the diphenyliodonium reagent suggests a crucial participation of this anion as well. This may include a direct interaction between the solvated acetate with the diboron reagent (state B) or a participation of its negative charge throughout the hydrogen-bonding network of the protic solvent (state C). For the latter scenario, the activation is reminiscent of the more common activation with methoxide base. Obviously, the anions from compounds 1a–c do not display sufficient basicity to induce activation of the diboron reagent. The importance of the polar protic solvent is evident from a comparison of a reaction in polar unprotic THF, which led to a significantly lower yield of 39% of 3a.22
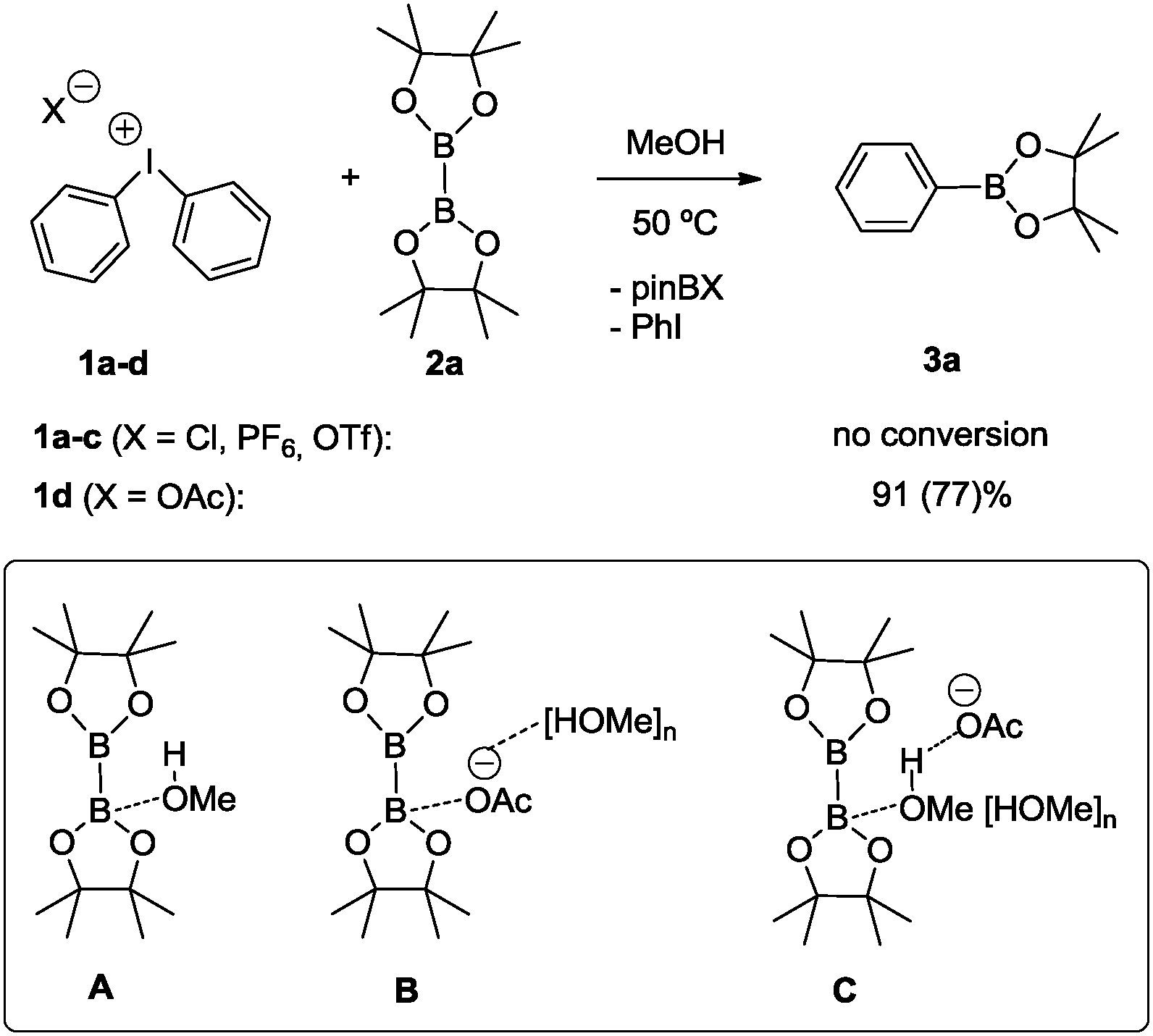 | ||
| Scheme 2 Borylation of diphenyliodonium salts: optimization with B2pin2. Reaction conditions: (C6H5)2I+X− (0.2 mmol), B2pin2 (0.3 mmol), 50 °C, 1.25 mL solvent, 24 h; average yield from two independent runs calculated by GC spectroscopy with mesitylene as the internal standard; the value in parenthesis refers to an isolated yield based on (C6H5)2IOAc. | ||
With the conceptual verification of the C–B bond formation in hand, the reaction was further extended to various diboron compounds for aryl-boron coupling (Scheme 3). For example, products 3b and 3c are conveniently generated by treatment between diphenyliodonium acetate and bis(neopentylglycolato) diboron (2b) and bis(hexyleneglycolato)diboron (2c), respectively, as in the case of the parent transformation with bis(pinacolato)diboron (2a). The same protocol was also applicable for the related bis(catecholato)diboron (2d) to furnish the coupling product 3d. Due to the notorious instability of the catecholboryl derivative, the boronic ester was transformed into phenol upon oxidative work-up.22 Finally, an excellent result was obtained using the mixed diboron reagent Bpin–Bdan (2e) (dan = 1,8-diaminonaphthalene),23 which underwent selective activation at the more electrophilic Bpin center.19 This promotes the transfer of the Bdan entity to selectively generate C–B coupling product 3e in 87% conversion and 52% isolated yield. There is only one precedence of borylation of aryl halides with Bpin–Bdan (2e), which required the presence of a palladium-XPhos catalyst under basic conditions.24
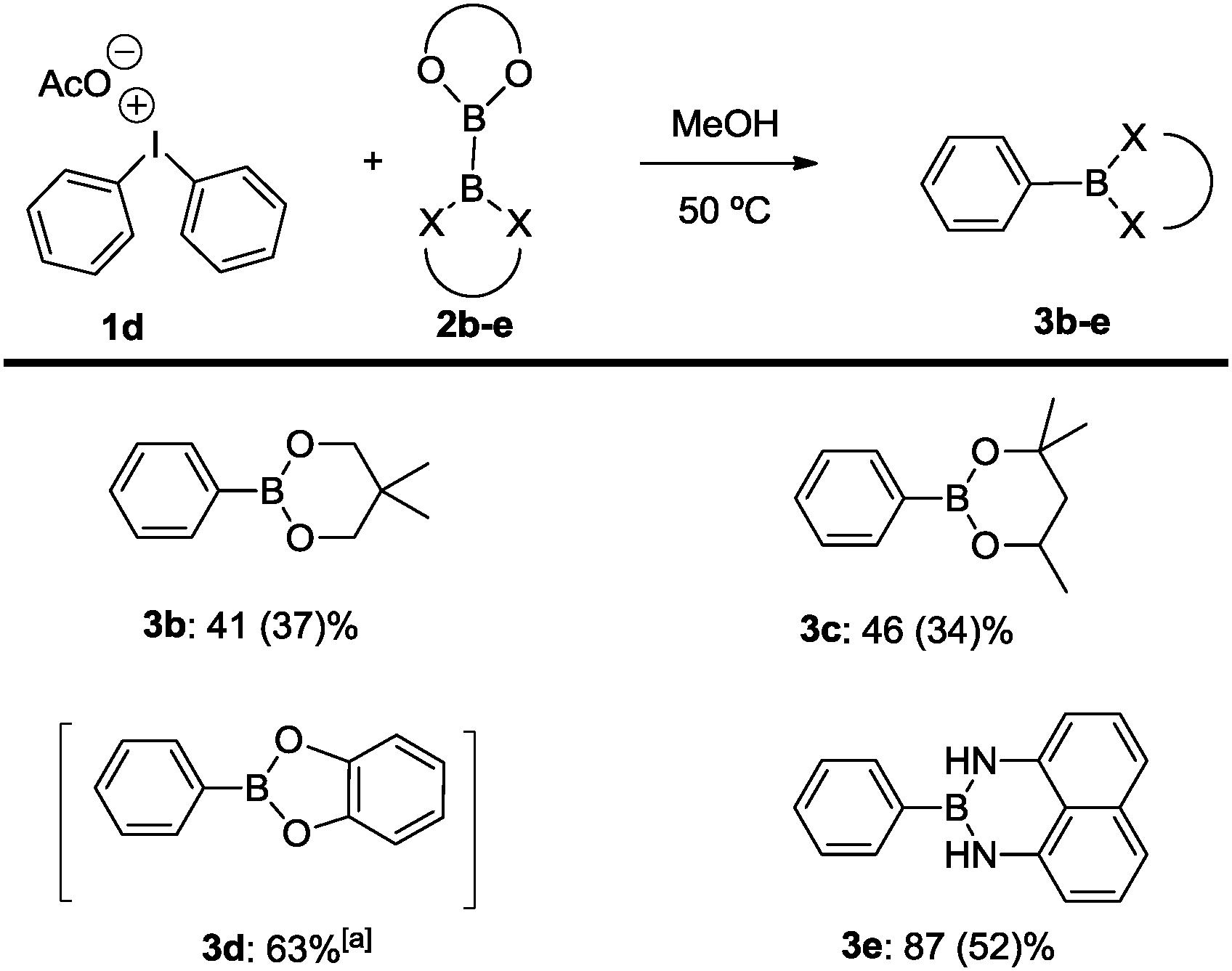 | ||
| Scheme 3 Borylation of diphenyliodonium salts: influence of the diboron reagent X = O, NH. Reaction conditions: (C6H5)2IOAc (0.2 mmol), diboron reagent (0.3 mmol), 50 °C, 1.25 mL methanol, 24 h; yield calculated by GC spectroscopy with mesitylene as the internal standard as an average of two reactions; isolated yields given in brackets calculated from (C6H5)2IOAc; [a] isolated yield of the corresponding alcohol after oxidative work up.22 | ||
The scope of the reaction has been evaluated for a number of symmetric diaryliodonium salts, which includes ortho-, meta and para-substitution patterns as well as higher substituted aromatic entities (Scheme 4). As to a general trend, electron releasing groups on the 4-substituted aryls of Ar2IOAc contribute to a higher percentage of borylated product formation (8a–10a) in comparison with the electron-withdrawing functional groups (products 4a–7a). In contrast, 3-substituted aryl groups in Ar2IOAc bearing electron-withdrawing substituents favor the borylation with the activated B2pin2 (products 11a–13a), while the corresponding diaryliodonium salt with 3-tolyl groups is transformed into coupling product 14a in moderate yield. As regularly encountered in arylboronates, yields can diminish during the purification step, being comparable to the most recent achievement in Zn catalyzed borylation of aryl halides.25
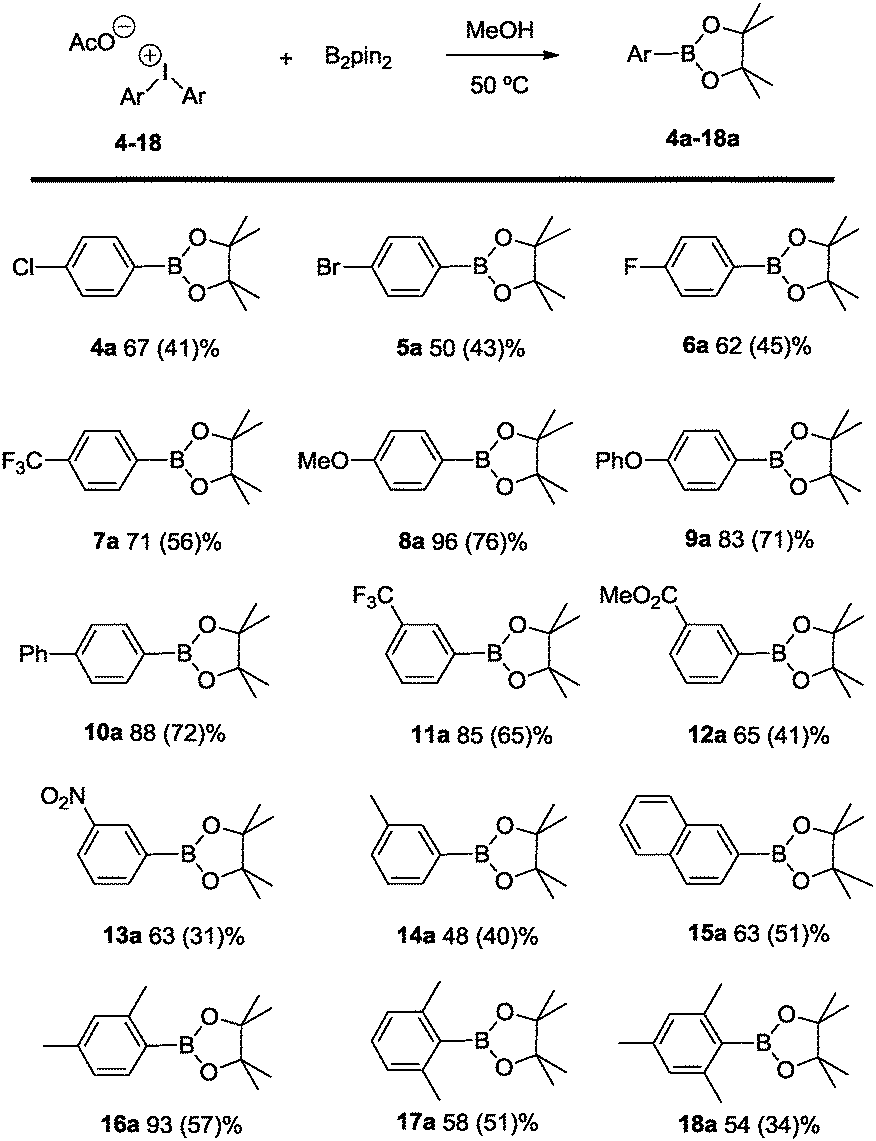 | ||
| Scheme 4 Borylation of diphenyliodonium salts: reaction scope with B2pin2. Reaction conditions: Ar2IOAc (0.2 mmol), B2pin2 (0.3 mmol), 50 °C, 1.25 mL methanol, 24 h; average yield from two independent runs calculated by GC spectroscopy with mesitylene as the internal standard; values in brackets refer to isolated yields based on Ar2IOAc. | ||
Interestingly, the highly substituted aryl(pinacolboronate) esters 15a–18a are quantitatively formed demonstrating that the present method provides great tolerance to formation of sterically encumbered aryl boronates.
With the aim to explore a selective mixed borylation we conducted two parallel strategies: (i) the reactivity of the novel non-symmetric diaryliodonium salts 19–23 with B2pin2 and (ii) the reactivity of electronically and sterically mixed diaryliodonium salts 19 and 20 with the mixed diboron reagent Bpin–Bdan (2e). Scheme 5 shows that sterically encumbered aryl boronates exert a higher degree of selective borylation when reacted with both symmetrical and mixed diboron compounds. Therefore, the diaryliodonium salt 19 reacts with 2a to give 7a and 19a in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, which is comparable to the reaction with 2e providing 7e and 19e in a ratio of 63:37. However, the higher the steric hindrance on one of the aryl groups, the more pronounced chemoselectivity favoring 3a is observed, regardless the electronic nature of the substituents on the neighboring aryl group. Interestingly, the reaction of 23 with 2e is conducted towards the formation of 3e and 3a in 9:1 ratio, showing that steric factors are predominant.
:1 ratio, which is comparable to the reaction with 2e providing 7e and 19e in a ratio of 63:37. However, the higher the steric hindrance on one of the aryl groups, the more pronounced chemoselectivity favoring 3a is observed, regardless the electronic nature of the substituents on the neighboring aryl group. Interestingly, the reaction of 23 with 2e is conducted towards the formation of 3e and 3a in 9:1 ratio, showing that steric factors are predominant.
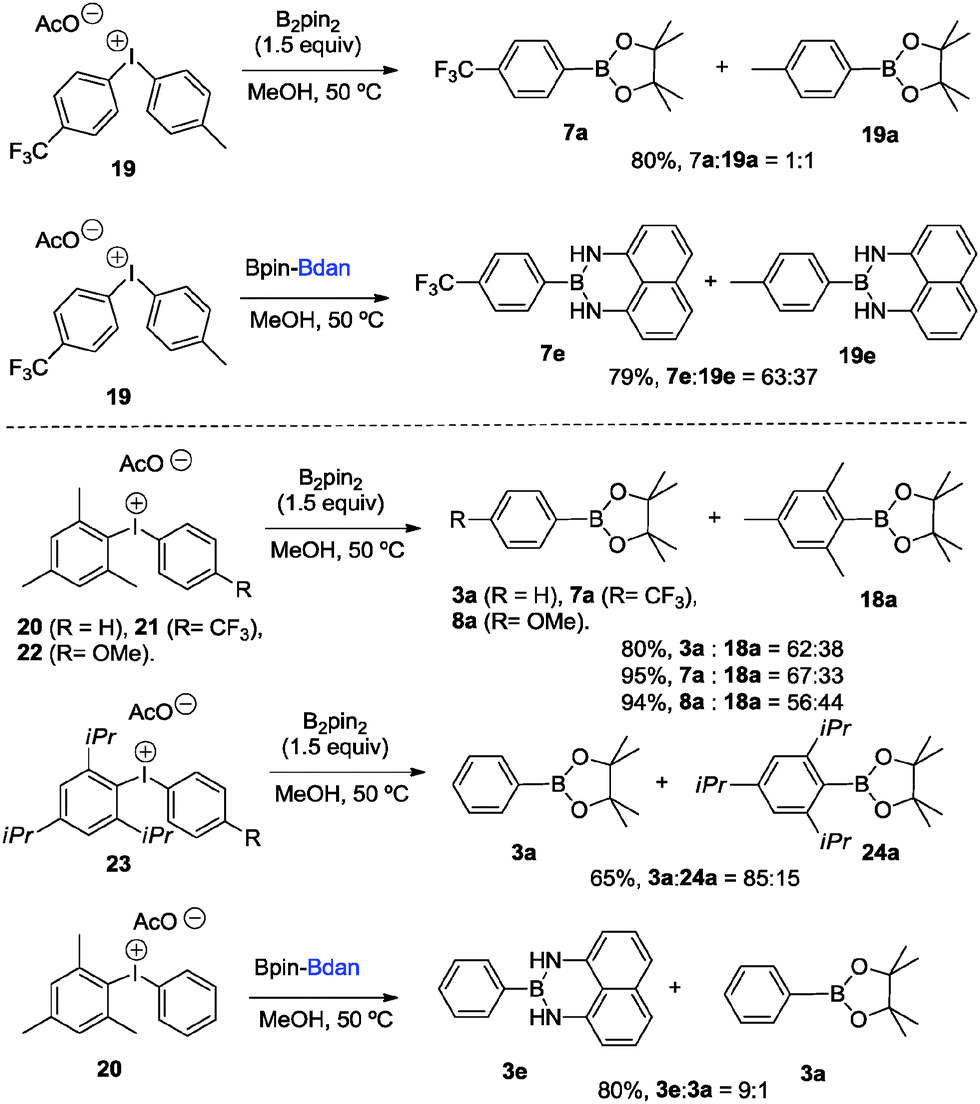 | ||
| Scheme 5 Borylation of non-symmetric diphenyliodonium salts. | ||
Finally, the successful development of the C–B bond forming reaction was extended to an in situ cross coupling reaction. Since the borylation reaction of the diaryliodonium salts 1 generates an equimolar amount of free aryl iodides, subjecting the crude reaction mixture to Suzuki–Miyaura cross-coupling should result in an overall diaryl synthesis. Such a process would make economic use of both of the aryl groups of the diaryliodonium precursor. Transformations of this kind are rare. Within this context, Beletskaya reported an exhaustive Suzuki–Miyaura coupling between diaryliodonium salts and sodium tetraphenylborate,26 Nachtsheim developed two sequential C–N bond forming events of cyclic diaryliodonium salts with anilines to yield N-arylated carbazoles27 and Greaney recently reported on the use of both aryl groups of diaryliodonium salts in 1,3-difunctionalization of indoles.28 Indeed, the anticipated reaction sequence comprising the C–B bond formation as a transient path to C–C coupling could be realized. After the borylation reaction, the solvent was changed from MeOH to toluene followed by addition of the palladium catalyst Pd(PPh3)4 and carbonate base (Scheme 6). By this, the mentioned product mixtures from C–B bond formation with symmetric diaryliodonium salts engage in the desired direct Suzuki–Miyaura coupling to provide the corresponding diaryl compounds 25–28 as the only C–C coupling products.
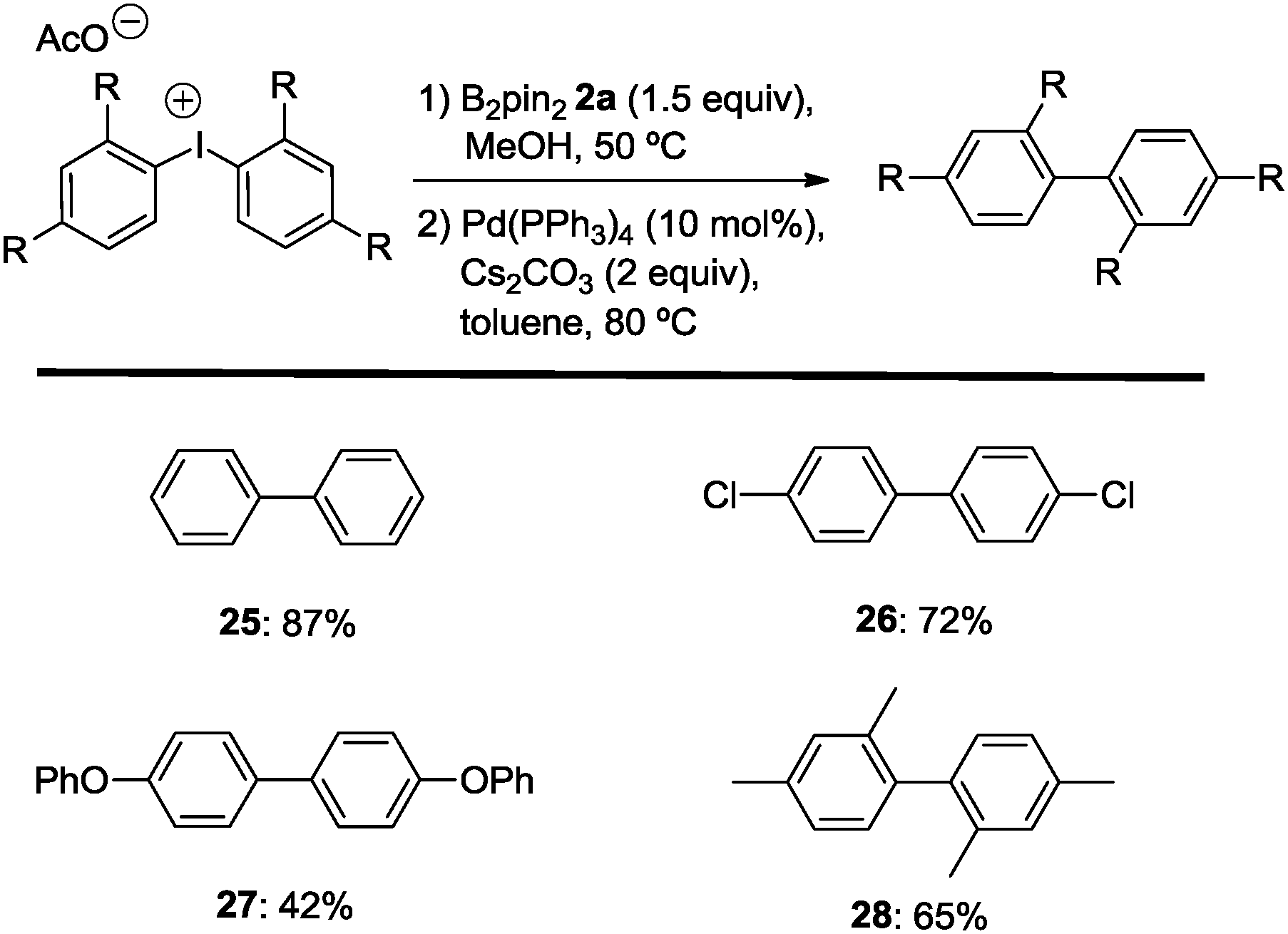 | ||
| Scheme 6 One pot cross coupling from diaryliodonium salts via B2pin2. Reaction conditions for activation: Ar2IOAc (0.2 mmol), B2pin2 (0.3 mmol), 50 °C, 1.25 mL MeOH, 24 h, reaction conditions for cross-coupling: Pd(PPh3)4 (10 mol%), Cs2CO3 (2 eq.), 80 °C, 2 mL toluene, 16 h. Yields refer to isolated yields based on Ar2IOAc. | ||
In summary, we have developed a new protocol for the metal-free formation of aryl–boron bonds. It employs readily available diaryliodonium acetates and commercially available diboron reagents, which in a methanol solution engage in direct aryl–boron bond formation through a formal umpolung of the electrophilic boron center. The reaction is selective, proceeds under mild conditions and does not require any metal reagent or other promoter. It opens a new methodological venue for the use of hypervalent diaryliodonium reagents in carbon–heteroatom bond formation. By simply adding a palladium source, the reaction can be directly expanded to biaryl synthesis through the cross coupling of symmetric diaryliodonium salts.
Financial support of this project was provided by MINECO (CTQ2014-56474R grant to K. M., CTQ2013-43395P grant to E. F., Severo Ochoa Excellence Accreditation 2014–2018 to ICIQ, SEV-2013-0319 and predoctoral FPI and FPU fellowships to N. M. and R. M. R.), and RedINTECAT CTQ2014-52974-REDC.
Notes and references
- (a) V. V. Zhdankin, Hypervalent Iodine Chemistry Preparation, Structureand Synthetic Applications of Polyvalent Iodine Compounds, Wiley, Chichester, 2013 Search PubMed; (b) P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123 CrossRef CAS PubMed; (c) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523 CrossRef CAS PubMed; (d) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS PubMed; (e) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS PubMed; (f) N. R. Deprez and M. S. Sanford, Inorg. Chem., 2007, 46, 1924 CrossRef CAS PubMed.
- Fluorination: (a) N. Ichiishi, A. J. Canty, B. F. Yates and M. S. Sanford, Org. Lett., 2013, 15, 5134 CrossRef CAS PubMed; (b) N. Ichiishi, A. F. Brooks, J. J. Topczewski, M. E. Rodnick, M. S. Sanford and P. J. H. Scott, Org. Lett., 2014, 16, 3224 CrossRef CAS PubMed , and references therein.
- Iodination: B. Hu, W. H. Miller, K. D. Neumann, E. J. Linstad and S. G. DiMagno, Chem. – Eur. J., 2015, 21, 6394 CrossRef CAS PubMed.
- Chlorination: M.-R. Zhang, K. Kumata, M. Takei, T. Fukumura and K. Suzuki, Appl. Radiat. Isot., 2008, 66, 1341 CrossRef CAS PubMed.
- Amination: (a) F. Tinnis, E. Stridfeldt, H. Lundberg, H. Adolfsson and B. Olofsson, Org. Lett., 2015, 17, 2688 CrossRef CAS PubMed; (b) J. Li and L. Liu, RSC Adv., 2012, 2, 10485 RSC , and references therein.
- Oxygenation: (a) B. Xiong, X. Feng, L. Zhu, T. Chen, Y. Zhou, C.-T. Au and S.-F. Yin, ACS Catal., 2015, 5, 537 CrossRef CAS; (b) R. Ghosh, E. Lindstedt, N. Jajalian and B. Olofsson, ChemistryOpen, 2014, 3, 54 CrossRef CAS PubMed , and references therein.
- Phosphorylation: Z.-D. Liu and Z.-C. Chen, Synthesis, 1993, 373 CrossRef CAS.
- Sulfonylation: (a) D. Wang, X. Yu, K. Zhao, L. Li and Y. Ding, Tetrahedron Lett., 2014, 55, 5739 CrossRef CAS PubMed; (b) N. Margraf and G. Manolikakes, J. Org. Chem., 2015, 80, 2582 CrossRef CAS PubMed , and references therein.
- Trifluoromethansulfination: S. C. Cullen, S. Shekhar and N. K. Nere, J. Org. Chem., 2013, 78, 12194 CrossRef CAS PubMed.
- Selenylation and telluration: Z.-D. Liu and Z.-H. Chen, Synth. Commun., 1993, 23, 2673 CrossRef CAS PubMed; J.-Z. You and Z.-C. Chen, Synthesis, 1992, 633 CrossRef.
- (a) Boronic Acids, ed. D. G. Hall, Wiley-VCH, Weinheim, 2011 Search PubMed; (b) Synthesis and Application of Organoboron Compounds, ed. E. Fernández and A. Whiting, Springer, 2015 Search PubMed.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) A. Suzuki, in Metal-catalyzed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998, pp. 49–97 Search PubMed; (c) J. C. H. Lee and D. G. Hall, in Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. De Meijere, S. Bräse and M. Oestreich, Wiley-VCH, Weinheim, 2014, vol. 1, pp. 65–132 Search PubMed; (d) A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412 RSC.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS PubMed.
- (a) T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508 CrossRef CAS; (b) T. Ishiyama, Y. Itoh, T. Kitano and N. Miyaura, Tetrahedron Lett., 1997, 38, 3447 CrossRef CAS; (c) G. A. Molander, S. L. J. Trice and S. D. Dreher, J. Am. Chem. Soc., 2010, 132, 17701 CrossRef CAS PubMed; (d) S. Kawamorita, H. Ohmiya, T. Iwai and M. Sawamura, Angew. Chem., Int. Ed., 2011, 50, 8363 CrossRef CAS PubMed; (e) G. A. Molander, S. L. J. Trice and S. M. Kennedy, Org. Lett., 2012, 14, 4814 CrossRef CAS PubMed.
- (a) C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350 CrossRef CAS PubMed; (b) K. Huang, D.-G. Yu, S.-F. Zheng, Z.-H. Wu and Z.-J. Shi, Chem. – Eur. J., 2011, 17, 786 CrossRef CAS PubMed; (c) T. Yamamoto, T. Morita, J. Takagi and T. Yamakawa, Org. Lett., 2011, 13, 5766 CrossRef CAS PubMed; (d) G. A. Molander, L. N. Cavalcanti and C. García-García, J. Org. Chem., 2013, 78, 6427 CrossRef CAS PubMed; (e) Y. Nagashima, R. Takita, K. Yoshida, K. Hirano and M. Uchiyama, J. Am. Chem. Soc., 2013, 135, 18730 CrossRef CAS PubMed; (f) S. K. Bose and T. B. Marder, Org. Lett., 2014, 16, 4562 CrossRef CAS PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed.
- J. Cid, J. J. Carbó and E. Fernández, Chem. – Eur. J., 2012, 18, 12794 CrossRef CAS PubMed.
- Activation of B2pin2 by alkoxyde: (a) A. Bonet, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2010, 49, 5130 CrossRef CAS PubMed; (b) A. Bonet, C. Pubill- Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2011, 50, 7158 CrossRef CAS PubMed; (c) C. Pubill-Ulldemolins, A. Bonet, C. Bo, H. Gulyás and E. Fernández, Chem. – Eur. J., 2012, 18, 1121 CrossRef CAS PubMed; (d) C. Solé, H. Gulyás and E. Fernández, Chem. Commun., 2012, 48, 3769 RSC; (e) C. Sole and E. Fernández, Angew. Chem., Int. Ed., 2013, 52, 11351 CrossRef CAS PubMed; (f) S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Mo, D. Qiu, M. S. Cheung, L. Dang, J. Wang and U. Radus, Chem. – Eur. J., 2015, 21, 7082 CrossRef CAS PubMed.
- Activation of BpinBdan by alkoxyde: (a) J. Cid, J. J. Carbó and E. Fernández, Chem. – Eur. J., 2014, 20, 3616 CrossRef CAS PubMed; (b) N. Mirallles, J. Cid, A. B. Cuenca, J. J. Carbó and E. Fernández, Chem. Commun., 2015, 51, 1693 RSC.
- For reviews on the field: (a) J. Cid, H. Gulyás, J. J. Carbó and E. Fernández, Chem. Soc. Rev., 2012, 41, 3558 RSC; (b) R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594 RSC.
- (a) Y. Nagashima, K. Hirano, R. Takita and M. Uchiyama, J. Am. Chem. Soc., 2014, 136, 8532 CrossRef CAS PubMed; (b) T. P. Blaisdell, T. C. Caya, L. Zhang, A. Sanz- Marco and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 9264 CrossRef CAS PubMed.
- See ESI† for details.
- N. Iwadate and M. Suginome, J. Am. Chem. Soc., 2010, 132, 2548 CrossRef CAS PubMed.
- L. Xu and P. Li, Chem. Commun., 2015, 51, 5656 RSC.
- (a) Y. Nagashima, R. Takita, K. Yoshida, K. Hirano and M. Uchiyama, J. Am. Chem. Soc., 2013, 135, 18730 CrossRef CAS PubMed; (b) S. K. Bose and T. B. Marder, Org. Lett., 2014, 16, 4562 CrossRef CAS PubMed.
- N. A. Bumagin, E. V. Luzikova, L. L. Sukhomlinova, T. P. Tolstaya and I. P. Beletskaya, Russ. Chem. Bull., 1995, 44, 385 CrossRef.
- S. Riedmüller and B. J. Nachtsheim, Beilstein J. Org. Chem., 2013, 9, 1202 CrossRef PubMed.
- S. G. Modha and M. F. Greaney, J. Am. Chem. Soc., 2015, 137, 1416 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc04944j |
| This journal is © The Royal Society of Chemistry 2015 |