
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide-based hydrogen sulphide-releasing gels†
Jennifer M.
Carter
,
Yun
Qian
,
Jeffrey C.
Foster
and
John B.
Matson
 *
*
Department of Chemistry and Macromolecules and Interfaces Institute, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: jbmatson@vt.edu
First published on 20th July 2015
Abstract
An aromatic peptide amphiphile was designed for delivery of the signaling gas H2S. The peptide self-assembled in water into nanofibers that gelled upon charge screening. The non-toxic gel slowly released H2S over 15 hours, and the presence of H2S in endothelial cells was verified using a fluorescent H2S probe.
Despite its reputation as a foul-smelling, toxic pollutant, hydrogen sulphide (H2S) is a vital biological signaling gas (gasotransmitter) produced endogenously by various enzymes.1 Within the past few years, efforts devoted to studying the biological roles of H2S, the enzymes that make it, and its therapeutic potential have expanded rapidly,2 along with related efforts to improve its detection in vitro and in vivo.3 While many biological studies on H2S are conducted using sulphide salts (NaHS and Na2S), injections of aqueous sulphide solutions lead to a rapid surge of H2S in the bloodstream followed by a rapid decline. To combat this uncontrolled delivery and more closely mimic the slow and sustained release of endogenous H2S, our group and others have reported on small molecules that release H2S with controllable kinetics.4 However, despite this progress on small molecule H2S donors, very few efforts have been made in the area of H2S-releasing materials.5
In addition to H2S, nitric oxide (NO) and carbon monoxide (CO) are also classified as gasotransmitters. The study of NO biology has benefitted greatly from NO-releasing materials, including polymers, gels, microparticles, and others.6 The localized release provided by these materials allows for site-specific delivery of the gas, which minimizes the required dosage and off-target effects that can result from systemic NO administration. In fact, NO-releasing materials have been studied widely in preclinical animal models for various applications.7 Additionally, CO releasing materials have also been reported, although they have not yet reached the stage of animal models.8 In contrast, few H2S-releasing materials have been prepared for potential use in therapy: two polymeric H2S donors5 and one H2S-releasing metal–organic framework.9 While polymers and other nanoparticle drug delivery systems may be useful for targeting specific tissues through systemic administration, H2S-releasing gels that can be localized at a site of interest are better suited as injectable biomaterials capable of filling a cavity or encapsulating cells while delivering precise doses of H2S to an area of interest.
We recently reported on a family of small molecule H2S donors based on the S-aroylthiooxime (SATO) functional group, which releases H2S in response to thiol functionality.4e SATOs are prepared by reaction of an S-aroyl thiohydroxylamine (Ar–C(O)S–NH2) (SATHA) with an aldehyde or ketone. H2S release is triggered by thiols such as cysteine, and release rates can be tuned by changing substituents on the SATHA ring, with half-lives of release in the range of 8–80 minutes.
In this report, we describe the preparation of a self-assembling peptide designed to form an H2S-releasing gel. Self-assembling peptide-based materials include a wide range of peptide motifs that form one-dimensional nanostructures in aqueous solution.10 Gel formation in the range of 0.5–2 wt% peptide can often be triggered by charge screening through physical entanglement of the extended nanostructures by pH changes or addition of salt. No chemical crosslinking is required, allowing for in situ gelation of the materials and shear-thinning behaviour. These qualities, along with their inherent biodegradability and minimal immunogenicity in most cases, make peptide-based gelators an attractive alternative to traditional polymeric hydrogels that require chemical crosslinking.
One class of self-assembling peptides is the aromatic peptide amphiphiles.11 Comprised of short oligopeptides with terminal aromatic components, these small molecule gelators can be designed to self-assemble in water into extended cylindrical micelles or nanofibers.12 The aromatic groups in aromatic peptide amphiphiles have traditionally served a structural role—none have included reactive groups designed to release a bioactive small molecule such as H2S.
Our design (Scheme 1) takes advantage of the inherent hydrophobicity of the SATO moiety to serve as both a structural component to drive self-assembly as well as a reactive functional group to release H2S. We envisioned that a SATO-functionalized aromatic peptide amphiphile could be designed to form 1-dimensional nanostructures capable of gelation. Additionally, we anticipated that the SATO functional groups would be buried in the hydrophobic core of the nanostructures, thereby limiting their access to the cysteine trigger required for decomposition and potentially extending their H2S-releasing capacity over hours to days.
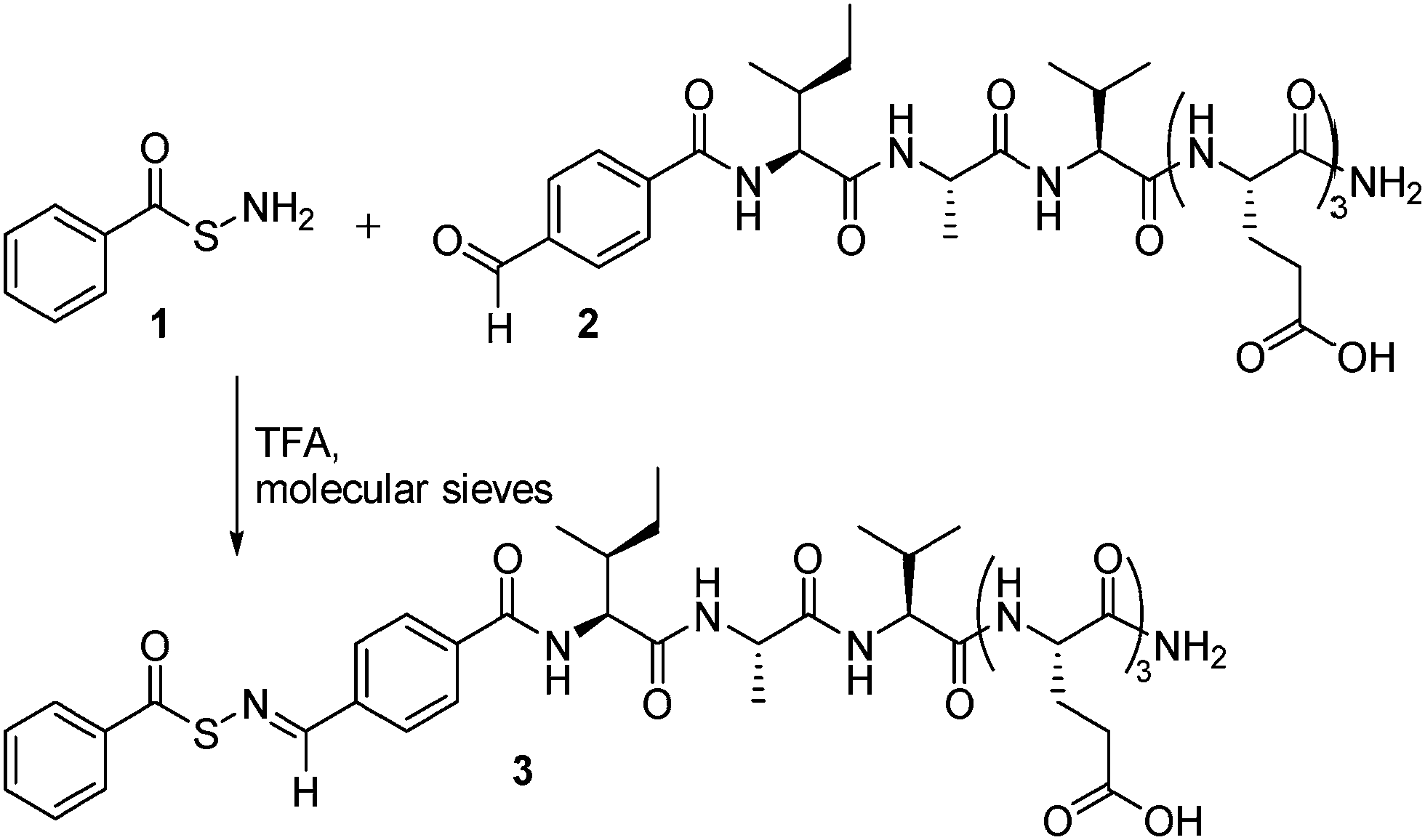 | ||
| Scheme 1 Synthesis of SATO-containing aromatic peptide amphiphile 3 from SATHA 1 and aldehyde-containing peptide 2. | ||
The peptide design we envisioned included a hydrophobic SATO group attached to a short peptide that facilitated assembly into 1-dimensional nanostructures. In addition to aromatic stacking interactions in the nanofiber core, many aromatic peptide amphiphiles also rely on β-sheet-type hydrogen bonding along the long axis of the nanostructure to stabilize the cylindrical assembly.13 Toward this end, the peptide IAVE3 was synthesized under standard solid phase conditions using Fmoc chemistry. The IAV region was designed based on the propensity of certain residues to self-pair in β-sheets found in natural proteins.14 Ala, Ile, and Val exhibit the highest tendency to self-pair; therefore, we incorporated these amino acids into the peptide sequence. The EEE sequence was included to provide the necessary hydrophilic/hydrophobic balance to achieve amphiphilicity. 4-Formylbenzoic acid (FBA) was added on resin under the same conditions as typical amino acid couplings, giving the modified peptide FBA-IAVE3 (2). Following cleavage and purification, peptide 2 was allowed to react with SATHA 1 in the presence of TFA, employing molecular sieves as a drying agent. Full conversion to peptide 3 was observed after 20 min by 1H NMR spectroscopy, and precipitation of the reaction mixture into CH2Cl2 afforded peptide 3, which was purified by preparative HPLC.
Transmission electron microscopy (TEM) was used to characterize the morphology of the self-assembled architecture of peptide 3 (Fig. 1A). Conventional TEM images revealed long, one-dimensional nanostructures with an average diameter of 10 nm. In contrast, TEM of peptide 2 (Fig. S5, ESI†) revealed predominately small spherical micelles around 10 nm in diameter and some larger spherical structures around 50 nm in diameter. No one-dimensional objects were observed.
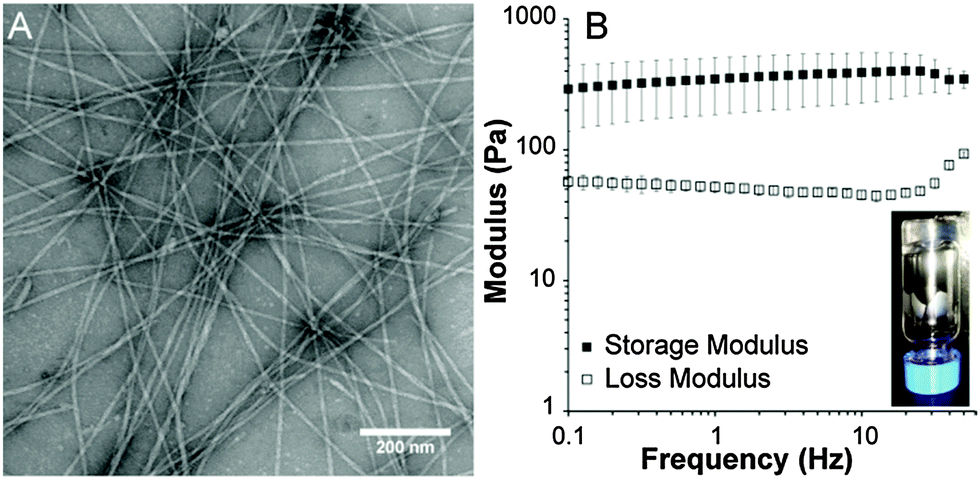 | ||
| Fig. 1 (A) Conventional TEM image of peptide 3 cast from water (stained with 2% uranyl acetate). (B) Frequency sweep rheology of peptide 3 at 1 wt% in water gelled with CaCl2. Inset shows photograph of self-supporting gel. | ||
Next, we examined whether peptide 3 was capable of forming a robust gel. Addition of aqueous CaCl2 solution to a 1 wt% solution of peptide 3 in water led to instantaneous gelation (Fig. 1B inset). As expected based on the TEM observations, addition of CaCl2 to a solution of peptide 2 did not result in gelation. The viscoelastic properties of the self-assembled peptide hydrogel prepared from a 1 wt% aqueous solution of peptide 3 were characterized by rheology (Fig. 1B). These experiments revealed that the gel was robust, with a storage modulus of 320 Pa. Both the storage and loss moduli for peptide 3 remained steady independent of frequency up to 50 Hz.
To evaluate the self-assembled structure of the nanofibers, we first measured the critical micelle concentrations (CMCs) of peptides 2 and 3 using the Nile red assay. Both peptides had CMC values of approximately 1 mg mL−1 (Fig. S6, ESI†). Next, we characterized peptide 3 by circular dichroism (CD), IR, UV-Vis, and fluorescence spectroscopies. For all four characterization techniques, experiments were performed at a concentration below the CMC (0.25 mg mL−1), above the CMC (10 mg mL−1), and on the gel (10 mg mL−1 gelled with CaCl2).
Self-assembling peptides often have strong intermolecular hydrogen bonds that align with the long axis of the self-assembled nanofibers, typically taking on a β-sheet confirmation. This type of assembly results in a minimum at 220 nm and maximum at 190 nm in the CD spectrum.15 Peptide 3 exhibits a minimum at 199 nm, which most resembles a random coil confirmation (Fig. 2A). Above the CMC, however, a minimum at 220 nm is observed. Unfortunately, the high concentration (10 mg mL−1) limited our ability to reliably collect data below 205 nm, so the expected maximum at 190 nm could not be observed. A similar spectrum is observed upon gelation at 10 mg mL−1, indicating that a β-sheet structure likely forms above the CMC and persists in the gel phase. Interestingly, a negative peak at 313 nm increases in intensity upon aggregation and gelation, likely indicating that the SATO chromophore resides in a chiral environment above the CMC of the peptide.
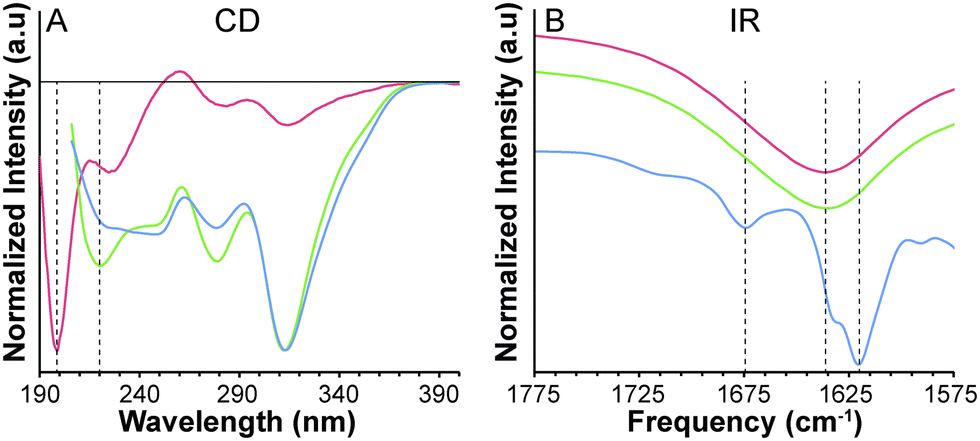 | ||
| Fig. 2 Characterization of peptide 3 at 0.25 mg mL−1 (red), 10 mg mL−1 (green) and 10 mg mL−1 in gel form (blue). (A) CD spectroscopy normalized to largest peak. The solid horizontal line represents the 0 line; the vertical dashed lines highlight the peaks indicative of random coil (199 nm) and β-sheet (220 nm) structures. (B) IR spectroscopy with y-axes shifted for clarity. The vertical dashed lines designate the amide I absorptions indicative of random coil (1637 cm−1) and β-sheet (1674 and 1620 cm−1) conformations. | ||
To further investigate the peptide secondary structure, IR spectroscopy was used to analyze the amide I band region (1600–1700 cm−1) of peptide 3. A strong absorption near 1630 cm−1 and a weaker absorption near 1680 cm−1 is typically observed when β-sheets are dominant.16 In contrast, absorptions for α-helices and random coils are both located near 1650 cm−1. Below the CMC, an absorption peak centered at 1637 cm−1 with no secondary absorption near 1680 cm−1 is observed for peptide 3. Above the CMC the absorption shifts to lower frequency at 1635 cm−1, but still without a secondary peak. Upon gelation, however, absorption peaks at 1620 cm−1 and 1674 cm−1 are present, indicative of a β-sheet structure.
UV-vis and fluorescence spectroscopy data (Fig. S7, ESI†) show absorptions at around 320 nm and emissions with peaks near 470 nm at all concentrations. No substantial redshift in fluorescence upon aggregation or gelation is observed. Taken together, the spectroscopy data reveal that peptide 3 adopts a random coil conformation below the CMC and increases in β-sheet character above the CMC and upon gelation. Extensive aromatic stacking is not present in these aggregates but may play a minor role in self-assembly.
The rate of H2S release from peptide 3 was measured using an H2S-selective microelectrode (Fig. 3). This method allows for real-time monitoring of the concentration of H2S in solution. Peptide 3 in solution released H2S more quickly than in gel form with an earlier peaking time (91 vs. 110 min) and a higher peak concentration (12.6 μM vs. 5.5 μM). Additionally, a period of slow initial release was observed for the gel, in contrast to the soluble peptide that immediately began releasing H2S upon addition of Cys. We attribute this induction period and overall slower release in gel form to limited diffusion of Cys into the gel. Importantly, measureable concentrations of H2S were released from the gel for over 15 h (Fig. S8, ESI†). No H2S release was observed in the absence of Cys.
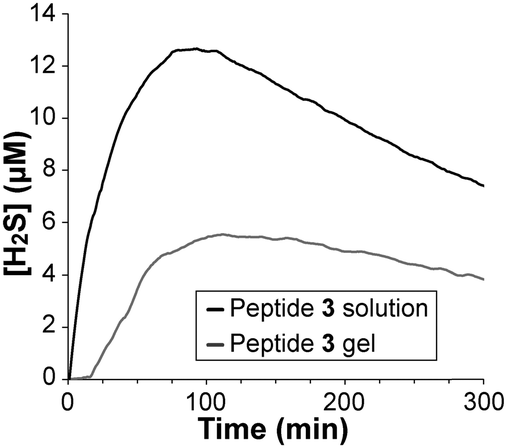 | ||
| Fig. 3 H2S release from peptide 3 (100 μM) in solution and as an aqueous gel (total of 100 μM in peptide 3). Both experiments were run in PBS containing 1 mM Cys. | ||
To assess whether peptide 3 could have potential use in vivo, a cytotoxicity study on mouse brain endothelial cells was conducted. Cytotoxicity of peptide 3 in solution was evaluated with and without Cys by measuring cell viability after 24 h (Fig. 4A). No toxicity was observed for the peptide 3 solution in either case up to 80 μM. Cytotoxicity of the peptide 3 gel at 1 wt% was also measured with and without Cys (Fig. 4B). Again, no significant toxicity was observed.
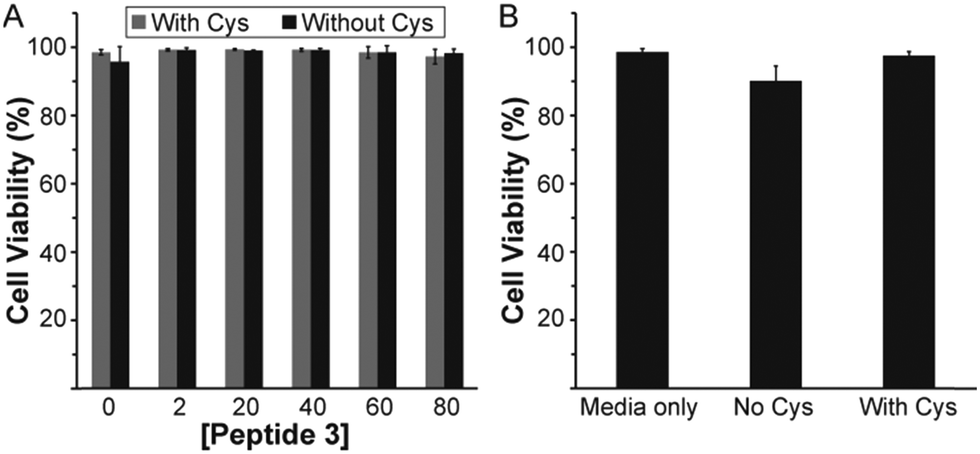 | ||
| Fig. 4 Cytotoxicity of peptide 3 on endothelial cells in basal media with or without Cys (1 mM). (A) In solution. (B) As 1 wt% aqueous gel. Viability was measured after 24 h by live/dead staining (calcein AM and ethidium homodimer). | ||
Lastly, H2S release from peptide 3 in the presence of endothelial cells was analyzed using a turn-on fluorescent probe selective for H2S (DT-OH).17 The addition of peptide 3 and Cys to cells pre-incubated with DT-OH showed strong fluorescence (Fig. 5). To confirm the fluorescence was a result of the H2S released from the peptide, several controls were performed: DT-OH only, DT-OH plus Cys, and DT-OH plus peptide 3 without Cys. Fluorescence was observed in neither the DT-OH only group nor the DT-OH + peptide 3 group. Weak signal was observed in the DT-OH + Cys group, which we attribute to a previously reported side reaction of similar probes with Cys.3b In future studies we will assess whether H2S release from peptide 3 occurs in solution followed by diffusion into cells, or whether peptide 3 is internalized into cells before thiol-triggered H2S release occurs.
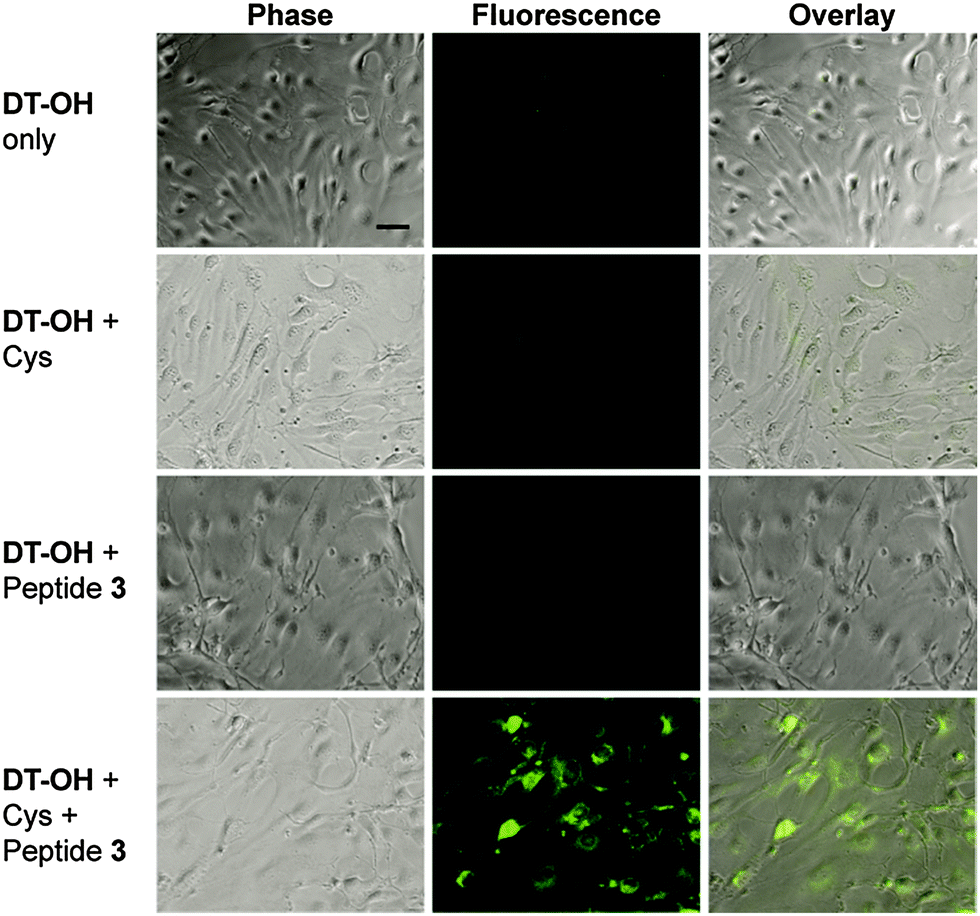 | ||
| Fig. 5 Phase, fluorescence, and overlay images showing strong fluorescence in endothelial cells preincubated with H2S probe DT-OH (5 μM) and then treated with peptide 3 (100 μM) and Cys (500 μM). Scale bar = 50 μm. | ||
In summary, we have designed and synthesized an H2S-releasing peptide that self-assembles into a robust hydrogel. The self-assembly of these peptides was guided primarily though β-sheet formation with a possible minor contribution from aromatic stacking interactions. Moreover, this peptide demonstrates that the aromatic component of self-assembling aromatic peptide amphiphiles can be designed to be both a structural unit as well as a reactive functional unit that decomposes to release a bioactive small molecule. These results also suggest that such gels may have therapeutic potential for localized H2S delivery. Investigations are currently underway in our laboratory to further explore the self-assembled structure of these peptides and to evaluate their applications as therapeutic H2S-releasing materials.
Notes and references
- (a) C. Szabo, Nat. Rev. Drug Discovery, 2007, 6, 917–935 CrossRef CAS PubMed; (b) H. Kimura, Antioxid. Redox Signaling, 2009, 12, 1111–1123 CrossRef PubMed; (c) R. Wang, Physiol. Rev., 2012, 92, 791–896 CrossRef CAS PubMed.
- (a) N. Shibuya, M. Tanaka, M. Yoshida, Y. Ogasawara, T. Togawa, K. Ishii and H. Kimura, Antioxid. Redox Signaling, 2009, 11, 703–714 CrossRef CAS PubMed; (b) A. Papapetropoulos, A. Pyriochou, Z. Altaany, G. Yang, A. Marazioti, Z. Zhou, M. G. Jeschke, L. K. Branski, D. N. Herndon, R. Wang and C. Szabó, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21972–21977 CrossRef CAS PubMed; (c) J. W. Calvert, W. A. Coetzee and D. J. Lefer, Antioxid. Redox Signaling, 2010, 12, 1203–1217 CrossRef CAS PubMed; (d) K. Kashfi and K. R. Olson, Biochem. Pharmacol., 2013, 85, 689–703 CrossRef CAS PubMed; (e) B. D. Paul, J. I. Sbodio, R. Xu, M. S. Vandiver, J. Y. Cha, A. M. Snowman and S. H. Snyder, Nature, 2014, 509, 96–100 CrossRef CAS PubMed; (f) N. Shibuya, S. Koike, M. Tanaka, M. Ishigami-Yuasa, Y. Kimura, Y. Ogasawara, K. Fukui, N. Nagahara and H. Kimura, Nat. Commun., 2013, 4, 1366 CrossRef PubMed.
- (a) A. R. Lippert, E. J. New and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 10078–10080 CrossRef CAS PubMed; (b) L. A. Montoya and M. D. Pluth, Chem. Commun., 2012, 48, 4767–4769 RSC; (c) B. Peng, W. Chen, C. Liu, E. W. Rosser, A. Pacheco, Y. Zhao, H. C. Aguilar and M. Xian, Chem. – Eur. J., 2014, 20, 1010–1016 CrossRef CAS PubMed; (d) X. Wang, J. Sun, W. H. Zhang, X. X. Ma, J. Z. Lv and B. Tang, Chem. Sci., 2013, 4, 2551–2556 RSC; (e) J. Cao, R. Lopez, J. M. Thacker, J. Y. Moon, C. Jiang, S. N. S. Morris, J. H. Bauer, P. Tao, R. P. Mason and A. R. Lippert, Chem. Sci., 2015, 6, 1979–1985 RSC.
- (a) Y. Zhao, H. Wang and M. Xian, J. Am. Chem. Soc., 2010, 133, 15–17 CrossRef PubMed; (b) Z. M. Zhou, M. V. Rekowski, C. Coletta, C. Szabo, M. Bucci, G. Cirino, S. Topouzis, A. Papapetropoulos and A. Giannis, Bioorg. Med. Chem., 2012, 20, 2675–2678 CrossRef CAS PubMed; (c) T. Roger, F. Raynaud, F. Bouillaud, C. Ransy, S. Simonet, C. Crespo, M. P. Bourguignon, N. Villeneuve, J. P. Vilaine, I. Artaud and E. Galardon, ChemBioChem, 2013, 14, 2268–2271 CrossRef CAS PubMed; (d) A. Martelli, L. Testai, V. Citi, A. Marino, I. Pugliesi, E. Barresi, G. Nesi, S. Rapposelli, S. Taliani, F. Da Settimo, M. C. Breschi and V. Calderone, ACS Med. Chem. Lett., 2013, 4, 904–908 CrossRef CAS PubMed; (e) J. C. Foster, C. R. Powell, S. C. Radzinski and J. B. Matson, Org. Lett., 2014, 16, 1558–1561 CrossRef CAS PubMed.
- (a) J. C. Foster and J. B. Matson, Macromolecules, 2014, 47, 5089–5095 CrossRef CAS; (b) U. Hasegawa and A. J. van der Vlies, Bioconjugate Chem., 2014, 25, 1290–1300 CrossRef CAS PubMed.
- (a) P. G. Parzuchowski, M. C. Frost and M. E. Meyerhoff, J. Am. Chem. Soc., 2002, 124, 12182–12191 CrossRef CAS PubMed; (b) N. A. Stasko and M. H. Schoenfisch, J. Am. Chem. Soc., 2006, 128, 8265–8271 CrossRef CAS PubMed; (c) M. R. Kapadia, L. W. Chow, N. D. Tsihlis, S. S. Ahanchi, J. W. Eng, J. Murar, J. Martinez, D. A. Popowich, Q. Jiang, J. A. Hrabie, J. E. Saavedra, L. K. Keefer, J. F. Hulvat, S. I. Stupp and M. R. Kibbe, J. Vasc. Surg., 2008, 47, 173–182 CrossRef PubMed.
- A. W. Carpenter and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC.
- (a) U. Hasegawa, A. J. van der Vlies, E. Simeoni, C. Wandrey and J. A. Hubbell, J. Am. Chem. Soc., 2010, 132, 18273–18280 CrossRef CAS PubMed; (b) N. E. Bruckmann, M. Wahl, G. J. Reiss, M. Kohns, W. Watjen and P. C. Kunz, Eur. J. Inorg. Chem., 2011, 4571–4577 CrossRef PubMed; (c) J. B. Matson, M. J. Webber, V. K. Tamboli, B. Weber and S. I. Stupp, Soft Matter, 2012, 8, 6689–6692 RSC; (d) A. E. Pierri, P. J. Huang, J. V. Garcia, J. G. Stanfill, M. Chui, G. Wu, N. Zheng and P. C. Ford, Chem. Commun., 2015, 51, 2072–2075 RSC.
- P. K. Allan, P. S. Wheatley, D. Aldous, M. I. Mohideen, C. Tang, J. A. Hriljac, I. L. Megson, K. W. Chapman, G. De Weireld, S. Vaesen and R. E. Morris, Dalton Trans., 2012, 41, 4060–4066 RSC.
- (a) S. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CrossRef CAS; (b) J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed; (c) D. J. Pochan, J. P. Schneider, J. Kretsinger, B. Ozbas, K. Rajagopal and L. Haines, J. Am. Chem. Soc., 2003, 125, 11802–11803 CrossRef CAS PubMed; (d) V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS PubMed; (e) J. P. Jung, A. K. Nagaraj, E. K. Fox, J. S. Rudra, J. M. Devgun and J. H. Collier, Biomaterials, 2009, 30, 2400–2410 CrossRef CAS PubMed.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- (a) Y. Zhang, H. Gu, Z. Yang and B. Xu, J. Am. Chem. Soc., 2003, 125, 13680–13681 CrossRef CAS PubMed; (b) D. M. Ryan, S. B. Anderson, F. T. Senguen, R. E. Youngman and B. L. Nilsson, Soft Matter, 2010, 6, 475–479 RSC; (c) S. Fleming, S. Debnath, P. Frederix, T. Tuttle and R. V. Ulijn, Chem. Commun., 2013, 49, 10587–10589 RSC.
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646–2651 CrossRef CAS PubMed.
- (a) T. S. Niwa and A. Ogino, J. Mol. Struct.: THEOCHEM, 1997, 419, 155–160 CrossRef CAS; (b) H.-H. Tsai, K. Gunasekaran and R. Nussinov, Structure, 2006, 14, 1059–1072 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
- A. Barth and C. Zscherp, Q. Rev. Biophys., 2002, 35, 369–430 CrossRef CAS.
- D. T. Shi, D. Zhou, Y. Zang, J. Li, G. R. Chen, T. D. James, X. P. He and H. Tian, Chem. Commun., 2015, 51, 3653–3655 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis and fluorescence spectroscopy data, CMC plots, additional TEM, rheology, and H2S-release data, experimental procedures, and characterization data for all new materials. See DOI: 10.1039/c5cc04883d |
| This journal is © The Royal Society of Chemistry 2015 |