
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New AIE-active dinuclear Ir(III) complexes with reversible piezochromic phosphorescence behaviour†
Guangfu
Li
a,
Xinyao
Ren
a,
Guogang
Shan
a,
Weilong
Che
a,
Dongxia
Zhu
*a,
Likai
Yan
a,
Zhongmin
Su
*a and
Martin R.
Bryce
*b
aInstitute of Functional Material Chemistry, Faculty of Chemistry, Northeast Normal University, Renmin Road 5268, Changchun 130024, P. R. China. E-mail: zhudx047@nenu.edu.cn; zmsu@nenu.edu.cn
bDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 16th July 2015
Abstract
Two new AIE-active dinuclear Schiff base Ir(III) complexes exhibit highly reversible piezochromic phosphorescence behaviour enabling the construction of a re-writable phosphorescence data recording device.
Phosphorescent transition metal complexes are widely exploited in various optoelectronic applications due to their rich excited-state properties, such as high luminescence quantum yields, long emission lifetimes, large Stokes shifts and high photo-stability compared to fluorescent dyes.1 However, there have been only limited studies focusing on the switching range of the phosphorescence emission of such materials in response to stimuli and their potential applications such as data recording and storage. Piezochromic materials, the emissions of which can be repeatedly switched between different colours under external pressure or mechanical grinding, have received considerable attention in the construction of optical data recording and storage devices.2 Recently, a series of organic small-molecules, metal complexes, liquid crystalline materials, polymers and metal–organic frameworks with intriguing piezochromic properties have been investigated.3 However, piezochromic phosphorescence has only rarely been observed in transition metal complexes and the relationship between changes in molecular assembly and their luminescence properties are not well understood. Moreover, similar to most conventional dyes, transition metal complexes usually suffer from low luminescence efficiency in the solid or aggregated state, due to aggregation-caused quenching (ACQ).4 This drawback in piezochromic materials significantly limits the real-world applications of these luminophores.
Aggregation-induced emission (AIE) in the solid or aggregated state, which is the opposite of ACQ, was first reported by Tang et al. in 2001.5 Recently, a number of pure organic fluorescent small molecules have been shown to simultaneously exhibit AIE and piezochromism.6 Very recently, Chi et al. proved that the positive effect of AIE on luminescence enhancement may provide a direction for the development of more efficient piezochromic materials.7 Therefore, AIE materials may become important alternative sources of piezochromic materials. However, phosphorescent luminophores with these dual properties are largely unexplored. Previous reports have focused on charged mono-iridium complexes with dendrimer-like or flexible alkyl chain substituents which exhibit piezochromism.8
Schiff base ligands play an important role in metal coordination chemistry, even after almost a century since their discovery, due to their facile synthesis, remarkable versatility and good solubility in common solvents.9 In general, compared with rigid ligands, a metal-coordinated Schiff base ligand can readily form intermolecular π–π or C–H⋯π interactions in the aggregated or crystal state because of the high flexibility of the imine unit. However, this structural flexibility can also induce relatively loose molecular packing, which might be easily collapsed by external pressure with a resulting effect on the HOMO–LUMO energy levels which would alter the luminescent properties.10 Thus, it is of interest to ask: “What will happen when Schiff base complexes are stimulated by external pressure?” Therefore, the development of a new class of Schiff base piezochromic materials with high luminescent efficiency is of fundamental importance in exploring the relationship between solid state structure and luminescence.
Inspired by this idea, herein, we describe two new dinuclear cationic Ir(III) complexes, [(2F-ppz)2Ir-(L1)-Ir(2F-ppz)2] [PF6]2 (1) and [(2F-ppz)2Ir-(L2)-Ir(2F-ppz)2] [PF6]2 (2) with Schiff base bridging ligands (L1) and (L2), respectively (Scheme 1). Their 1H NMR spectra, photophysical properties, powder X-ray diffraction, single-crystal X-ray structure (for 1) and differential scanning calorimetric (DSC) data are presented. The results obtained demonstrate that both complexes 1 and 2 are AIE-active and simultaneously show piezochromism and vapochromic phosphorescence. We conclude that the flexible bridging ligands play a significant role in achieving these combined properties. Most importantly, the highly reversible piezochromic behaviour makes both complexes competitive candidates for practical applications. Indeed, we have shown that complex 2 provides a fast-responding re-writable phosphorescence data recording device.
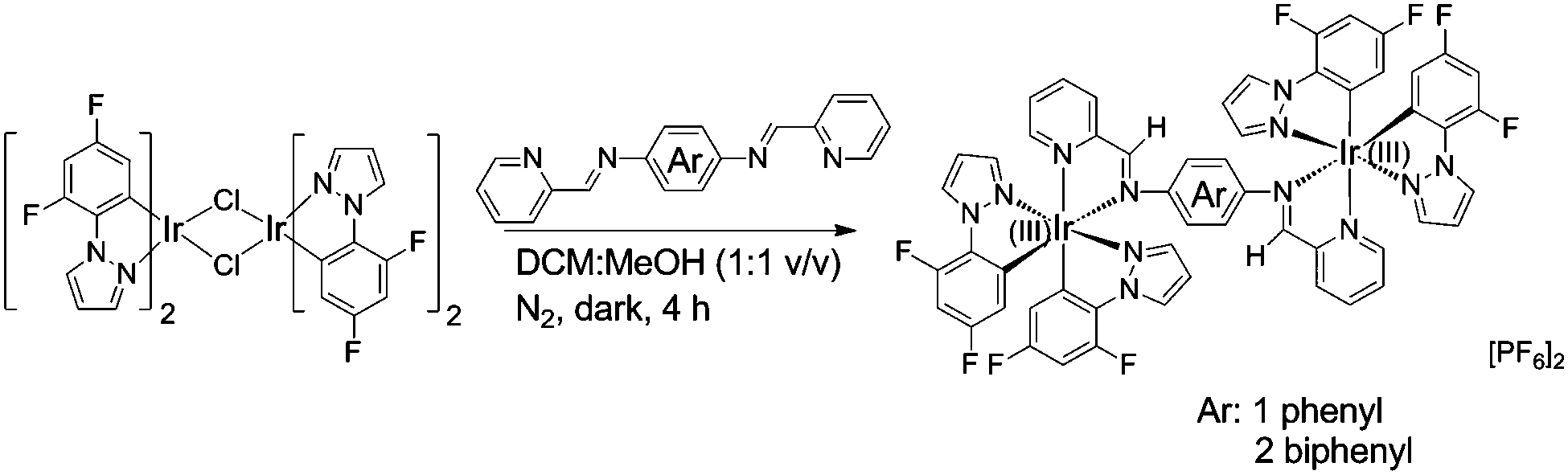 | ||
| Scheme 1 Chemical structures of the complexes, with bridging phenyl (1) and 4,4′-biphenyl (2) units. | ||
The UV/vis absorption and emission spectra of complexes 1 and 2 in degassed solution (CH3CN) are depicted in Fig. 1a. Upon photoexcitation, complexes 1 and 2 are almost non-emissive in pure CH3CN solution. However, the powdered samples of 1 and 2 exhibit intense phosphorescence at room temperature, with Φem 0.31 and 0.24, respectively (Table S1, ESI†). Such strong luminescence in the solid state is a requirement for luminescence switching in experiments of piezochromic behaviour (see below).
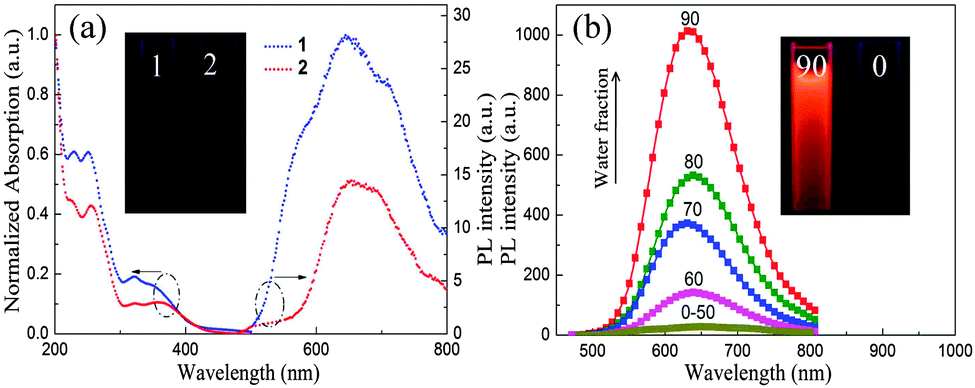 | ||
| Fig. 1 (a) Absorption and emission spectra of complexes 1 and 2 in acetonitrile solution at room temperature. Inset: emission image of complexes 1 and 2 in pure acetonitrile solution under 365 nm UV illumination; (b) emission spectra of complex 1 in CH3CN–water mixtures with different water fractions (0–90% v/v) at room temperature. Inset: emission image of complex 1 in pure acetonitrile solution and CH3CN–water mixture (fw = 90%) under 365 nm UV illumination. | ||
To probe the AIE of complexes 1 and 2, their photoluminescence (PL) spectra in CH3CN–H2O mixtures with various water contents were obtained (Fig. 1b and Fig. S1, ESI†). The phosphorescence intensity of both complexes is dramatically enhanced when the water fraction reached 60%. Furthermore, transmission electron microscopy (TEM) and electron diffraction (ED) experiments indicated that amorphous molecular aggregates are formed in the mixtures (Fig. S2, ESI†).8a The results confirm that complexes 1 and 2 are AIE active. To gain further understanding of the unusual solid-state emission properties, the geometry of complex 1 was optimized by referring to the X-ray diffraction data, and the electronic properties of the frontier orbitals were studied in the solution and the solid state structures using density functional theory (DFT) methods (Fig. 2).
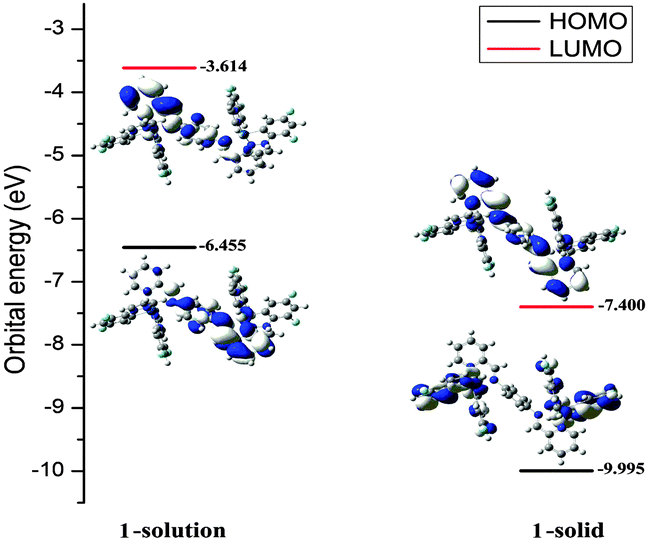 | ||
| Fig. 2 Molecular orbital diagrams, HOMO and LUMO energies for complex 1 at its S0 optimized geometries in solution state and solid state. | ||
In the solution state of complex 1, both the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) mainly reside on the Schiff base bridging ligand. In essence, this Intra-Ligand Charge Transfer (ILCT) excited state is harmful for luminescence.11 Moreover, based on the crystallographic analysis and DFT data for 1 there are large structural distortions in the T1 geometry compared to the S0 geometry. These distortions induce an excited state relaxation and may result in an effective pathway for nonradiative decay, which can explain the weak emission in the solution state (Table S3, ESI†).12 By comparison, in the solid state, obvious Metal-to-Ligand Charge Transfer (MLCT) and Ligand-to-Ligand Charge Transfer (LLCT) excited states exist and these will favour more radiative processes than ILCT excited states and thereby greatly enhance the luminescence in the solid state.13
Under UV light irradiation the as-prepared powders 1 and 2 exhibit yellow and orange phosphorescence at λmax 612 nm and 627 nm, respectively (Fig. S3, ESI†). These samples are hereafter referred to as P1 and P2. Interestingly, grinding both complexes on quartz plates (to give samples referred to as G1 and G2, respectively) induced a red-shift of the emission by ca. 20 nm to λmax 635 nm and 648 nm, respectively (Fig. S4 and S5, ESI†), clearly visible to naked eyes. Evidently, both P1 and P2 exhibit pressure-induced piezochromic behaviour. To investigate the reversibility of this behaviour, G1 and G2 were exposed to CH2Cl2 solvent vapour which caused the emission spectra to revert to the original spectra of P1 and P2 within a few seconds (Fig. S5, ESI†), demonstrating a vapochromic effect because of vapour-induced recrystallization.14
By exploiting the piezochromic and vapochromic responses of complex 2, a re-writable phosphorescence data recording device has been constructed (Fig. 3). The procedure is as follows. When the as-prepared ground powder G2 is carefully spread on a filter paper using a porcelain pestle to make a thin film, it emits red light upon excitation with a UV lamp. Then a letter “r” was written on the ‘paper’ using a ‘pen’ (made from a glass pipette) with CH2Cl2 vapour as the ‘ink’, and an orange-emitting symbol with large colour contrast is observed. Erasing the letter “r” by grinding reinstalls the original red background. A new letter “I” can now be written on the paper and erased using the same method described above. The writing and erasing processes can be repeated many times.
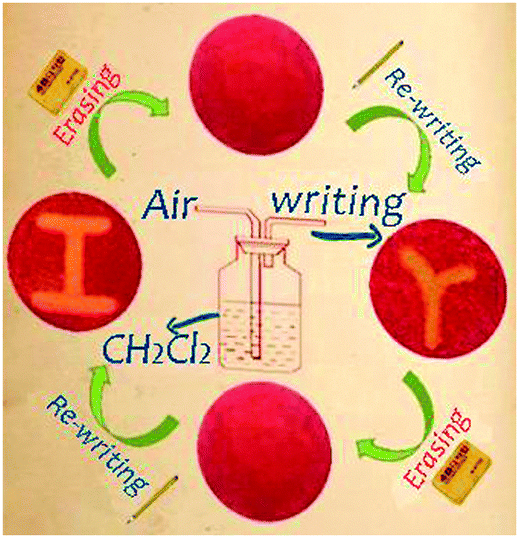 | ||
| Fig. 3 A phosphorescence re-writable data recording device based on mechanochromic and vapochromic phosphorescence of complex 2. | ||
To understand the origin of this reversible piezochromic behaviour of complexes 1 and 2, NMR spectroscopy, X-ray crystallographic analysis, time-resolved emission decay, powder X-ray diffraction (PXRD) combined with differential scanning calorimetry (DSC) studies were performed. 1H NMR spectra showed similar peak shapes and chemical shift values for both samples P2 and G2 (Fig. S11, ESI†). This result proves that no chemical reaction occurs during the grinding process. Therefore, we conclude that piezochromic behaviour is caused by physical processes, such as changing the intermolecular interactions and/or the mode of the molecular packing.
The single crystal packing structure of complex 1 reveals an obvious intermolecular π–π interaction between the neighbouring pyrazole rings which induces a face-to-face aggregation (Fig. 4). This packing might be easily modified by external mechanical pressure resulting in increased molecular conjugation, thereby facilitating a red-shift of the PL spectrum.15 The excited-state lifetimes (τ) for the as-synthesized samples P1 (0.18 μs) and P2 (0.17 μs) are significantly different from those of ground samples G1 (0.22 μs) and G2 (0.20 μs): the grinding results in an increase of τ, indicating that the mode of solid-state molecular packing and/or the intermolecular interactions are altered after grinding which weakens the intermolecular π–π interactions.16 For G1 and G2 the excited-state lifetimes (τ) of heated or CH2Cl2 fumed samples and as-synthesized samples are almost identical (Table S2, ESI†) confirming that they are the same species.
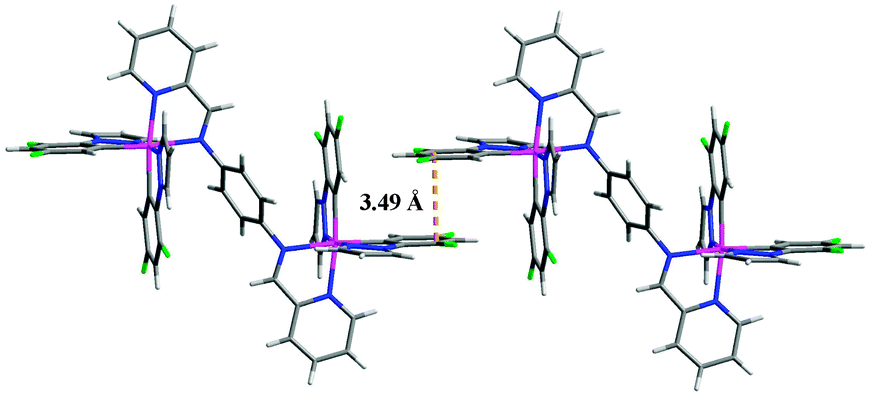 | ||
| Fig. 4 Interaction between two molecules of complex 1 in the X-ray crystal structure. The PF6− anions are omitted for clarity. | ||
The powder X-ray diffraction (PXRD) patterns (Fig. 5a and b) are consistent with the single-crystal diffraction data. The intense and sharp reflection peaks demonstrate that P1 and P2 are well-ordered aggregates. In contrast, the ground samples G1 and G2 show very weak and broad diffraction signals, indicating their amorphous states. After heating or exposing the ground samples to CH2Cl2 some sharp diffraction peaks reappeared. Thus, piezochromic reversibility for complexes 1 and 2 is ascribed to crystallization and amorphization upon the grinding–heating (or vapour exposure) process. Small differences in the patterns between P and Heated G and Fumed G samples could be ascribed to different aggregated states after treatment. In addition, upon heating G1 and G2 to 350 °C, the DSC traces exhibited a clear broad exothermic recrystallization peak at ca. 280 °C and 290 °C, respectively (Fig. 5c and d). This peak is at a similar temperature at which thermal recrystallization begins to take place. When G1 and G2 were heated at 290 °C for 1 min, the emission colours also reverted to their original colours. Upon further grinding of the heated samples again, a highly reproducible red-shift of the emission again occurred. This piezochromic behaviour of complexes 1 and 2 was shown to be reversible for many cycles (Fig. 5e and f).
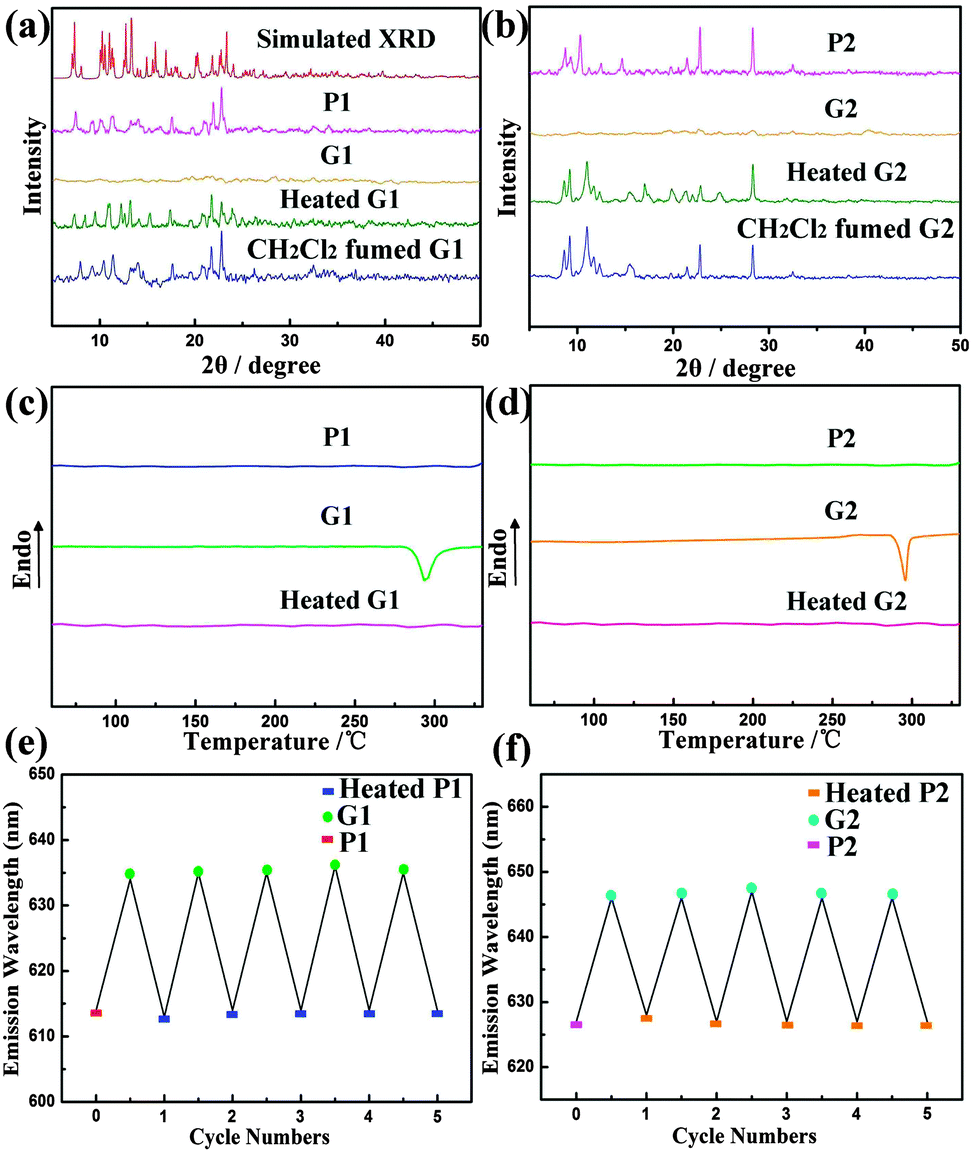 | ||
| Fig. 5 Powder X-ray diffraction patterns (a) and (b) and the DSC traces (c) and (d) of the corresponding samples; repeated cycles of the piezochromism (e) and (f). | ||
In summary, two new AIE-active cationic dinuclear Ir(III) complexes which show highly reversible piezochromic phosphorescence are reported. It is proposed that the flexible imine units of the Schiff base bridging ligand play an important role in achieving AIE, while the phenylpyrazole groups lead to simultaneous piezochromism. Developing new Ir(III) complexes with versatile and flexible bridging ligands holds great promise as a new strategy to achieve highly efficient piezochromic and AIE phosphorescence materials in the future.
The work in China was funded by NSFC (No. 51203017, No. 51473028 and No. 21303012), the key scientific and technological project of Jilin province (20150204011GX). EPSRC funded the work in Durham.
Notes and references
- (a) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS; (b) S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS PubMed; (c) M. Mydlak, C. Bizzarri, D. Hartmann, W. Sarfert, G. Schmid and L. De Cola, Adv. Funct. Mater., 2010, 20, 1812 CrossRef CAS PubMed; (d) H. Sasabe, J.-i. Takamatsu, T. Motoyama, S. Watanabe, G. Wagenblast, N. Langer, O. Molt, E. Fuchs, C. Lennartz and J. Kido, Adv. Mater., 2010, 22, 5003 CrossRef CAS PubMed; (e) V. W.-W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579 RSC; (f) S. Ladouceur and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 2895 Search PubMed; (g) A. M. Bünzli, E. C. Constable, C. E. Housecroft, A. Prescimone, J. A. Zampese, G. Longo, L. Gil-Escrig, A. Pertegás and H. J. Bolink, Chem. Sci., 2015, 6, 2843 RSC; (h) A. Auffrant, A. Barbieri, F. Barigelletti, J. Lacour, P. Mobian, J.-P. Collin, J.-P. Sauvage and B. Ventura, Inorg. Chem., 2007, 46, 6911 CrossRef CAS PubMed.
- (a) M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334 CrossRef CAS PubMed; (b) M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755 CrossRef CAS PubMed; (c) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878 RSC; (d) Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782 CrossRef CAS PubMed; (e) A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546 CrossRef CAS PubMed; (f) W. Z. Yuan, Y. Tan, Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. Feng, H. H. Y. Sung, Y. Lu, I. D. Williams, J. Z. Sun, Y. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837 CrossRef CAS PubMed.
- (a) H. Bi, D. Chen, D. Li, Y. Yuan, D. Xia, Z. Zhang, H. Zhang and Y. Wang, Chem. Commun., 2011, 47, 4135 RSC; (b) C. Y. K. Chan, Z. Zhao, J. W. Y. Lam, J. Liu, S. Chen, P. Lu, F. Mahtab, X. Chen, H. H. Y. Sung, H. S. Kwok, Y. Ma, I. D. Williams, K. S. Wong and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 378 CrossRef CAS PubMed; (c) Y. Dong, J. Zhang, X. Tan, L. Wang, J. Chen, B. Li, L. Ye, B. Xu, B. Zou and W. Tian, J. Mater. Chem. C, 2013, 1, 7554 RSC; (d) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175 CrossRef CAS PubMed; (e) S. Xu, T. Liu, Y. Mu, Y. F. Wang, Z. Chi, C. C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874 CrossRef CAS PubMed; (f) S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675 CrossRef CAS PubMed; (g) X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376 RSC.
- (a) B. Xu, J. He, Y. Mu, Q. Zhu, S. Wu, Y. Wang, Y. Zhang, C. Jin, C. Lo, Z. Chi, A. Lien, S. Liua and J. Xu, Chem. Sci., 2015, 6, 3236 RSC; (b) B. Xu, M. Xie, J. He, B. Xu, Z. Chi, W. Tian, L. Jiang, F. Zhao, S. Liu, Y. Zhang, Z. Xu and J. Xu, Chem. Commun., 2013, 49, 273 RSC.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- (a) T. Butler, W. A. Morris, J. Samonina-Kosicka and C. L. Fraser, Chem. Commun., 2015, 51, 3359 RSC; (b) Q. Lu, X. Li, J. Li, Z. Yang, B. Xu, Z. Chi, J. Xu and Y. Zhang, J. Mater. Chem. C, 2015, 3, 1225 RSC; (c) R. Misra, T. Jadhav, B. Dhokale and S. M. Mobin, Chem. Commun., 2014, 50, 9076 RSC; (d) G. F. Zhang, H. Wang, M. P. Aldred, T. Chen, Z. Q. Chen, X. Meng and M. Q. Zhu, Chem. Mater., 2014, 26, 4433 CrossRef CAS.
- B. Xu, J. He, Y. Mu, Q. Zhu, S. Wu, Y. Wang, Y. Zhang, C. Jin, C. Lo, Z. Chi, A. Lien, S. Liua and J. Xu, Chem. Sci., 2015, 6, 3236 RSC.
- (a) G. G. Shan, H. B. Li, J. S. Qin, D. X. Zhu, Y. Liao and Z. M. Su, Dalton Trans., 2012, 41, 9590 RSC; (b) G. G. Shan, H. B. Li, H. Z. Sun, D. X. Zhu, H. T. Cao and Z. M. Su, J. Mater. Chem. C, 2013, 1, 1440 RSC; (c) G. G. Shan, H. B. Li, H. T. Cao, D. X. Zhu, P. Li, Z. M. Su and Y. Liao, Chem. Commun., 2012, 48, 2000 RSC; (d) G. G. Shan, H. B. Li, D. X. Zhu, Z. M. Su and Y. Liao, J. Mater. Chem. C, 2012, 22, 12736 RSC.
- (a) L. Sacconi, Coord. Chem. Rev., 1966, 1, 126 CrossRef CAS; (b) M. Nath and S. Goyal, Main Group Met. Chem., 1996, 19, 75 CrossRef CAS; (c) M. Nath and P. K. Saini, Dalton Trans., 2011, 40, 7077 RSC.
- S. P. Anthony, ChemPlusChem, 2012, 77, 518 CrossRef CAS PubMed.
- (a) S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Inorg. Chem., 2001, 40, 1704 CrossRef CAS PubMed; (b) C. H. Shin, J. O. Huh, S. J. Baek, S. K. Kim, M. H. Lee and Y. Do, Eur. J. Inorg. Chem., 2010, 3642 CrossRef CAS PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
- K. Huang, H. Wu, M. Shi, F. Li, T. Yi and C. Huang, Chem. Commun., 2009, 1243 RSC.
- J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956 CrossRef CAS PubMed.
- X. Zhang, Z. Chi, H. Li, B. Xu, X. Li, W. Zhou, S. Liu, Y. Zhang and J. Xu, Chem. – Asian J., 2011, 6, 808 CrossRef CAS PubMed.
- S. Mizukami, H. Houjou, K. Sugaya, E. Koyama, H. Tokuhisa, T. Sasaki and M. Kanesato, Chem. Mater., 2005, 17, 50 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, figures and procedures for the DFT calculations. CCDC 1046679. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc04850h |
| This journal is © The Royal Society of Chemistry 2015 |