
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
β-Mannosylation with 4,6-benzylidene protected mannosyl donors without preactivation†
Mads
Heuckendorff
,
Pernille Sørensen
Bols
,
Collin Bartholomew
Barry
,
Tobias Gylling
Frihed
 ,
Christian Marcus
Pedersen
* and
Mikael
Bols
*
,
Christian Marcus
Pedersen
* and
Mikael
Bols
*
Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark. E-mail: cmp@chem.ku.dk; bols@chem.ku.dk
First published on 13th July 2015
Abstract
Mannosylations with benzylidene protected mannosyl donors were found to be β-selective even when no preactivation was performed. It was also found that the kinetic β-product in some cases anomerizes fast to the thermodynamically favored α-anomer under typical reaction conditions.
Chemical glycosylation, a reaction for the synthesis of oligosaccharides and other biologically or medicinally crucial glycosides, is a very important reaction.1 Many excellent glycosylation protocols that lead to high yield and stereocontrol have been developed, even when alcohol and the glycosylation agent are used in equimolar amounts.2 Nevertheless, stereochemistry is always an issue in glycosylation reactions and this simple fact leaves the impression that they are largely SN1-type reactions.2d,3 It was therefore remarkable when the Crich laboratory discovered that β-mannosides, a stereochemical pattern that is notoriously difficult to obtain by direct glycosylation, could be obtained directly when a 4,6-benzylidene protective group was present in the mannosyl donor.4,5 It was also remarkable that a special pre-activation procedure was critical for β-selectivity: when the sulfoxide donor 1 was pre-activated with triflic anhydride at −78 °C before the addition of the acceptor, β-selectivity (β-mannoside 3) was obtained (Scheme 1), but if alcohol was present when triflic anhydride was added α-mannoside 4 was formed.6 These observations, together with the low temperature observation of an α-triflate 2 upon pre-activation, led to the proposal that the β-manno selectivity is a result of an SN2 substitution (Scheme 1),7,8 albeit several papers have reported that preactivation is not necessary.9 We were therefore surprised when we recently performed mannosylation reactions with a 4,6-silylene protected thiomannoside 5 and found that β-mannoside 7 was formed by simple direct activation of the thioglycoside with NIS/TfOH (cat.) in the presence of an acceptor (Scheme 2) – this worked even at room temperature, regardless of the anomeric configuration of the thiomannoside. This indicates that the oxocarbenium ion 6 was the glycosylating species and was attacked from the β-face.10
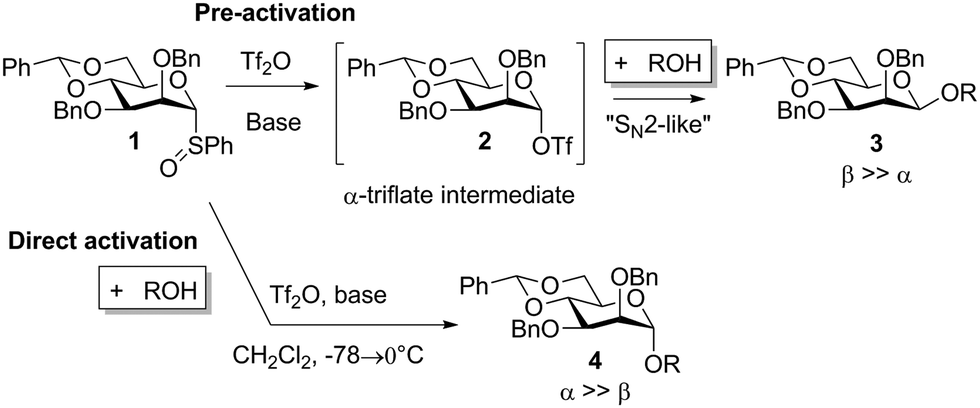 | ||
| Scheme 1 The crucial effect of the order of addition order on stereoselectivity in mannosylation reactions observed (ref. 4 and 6). | ||
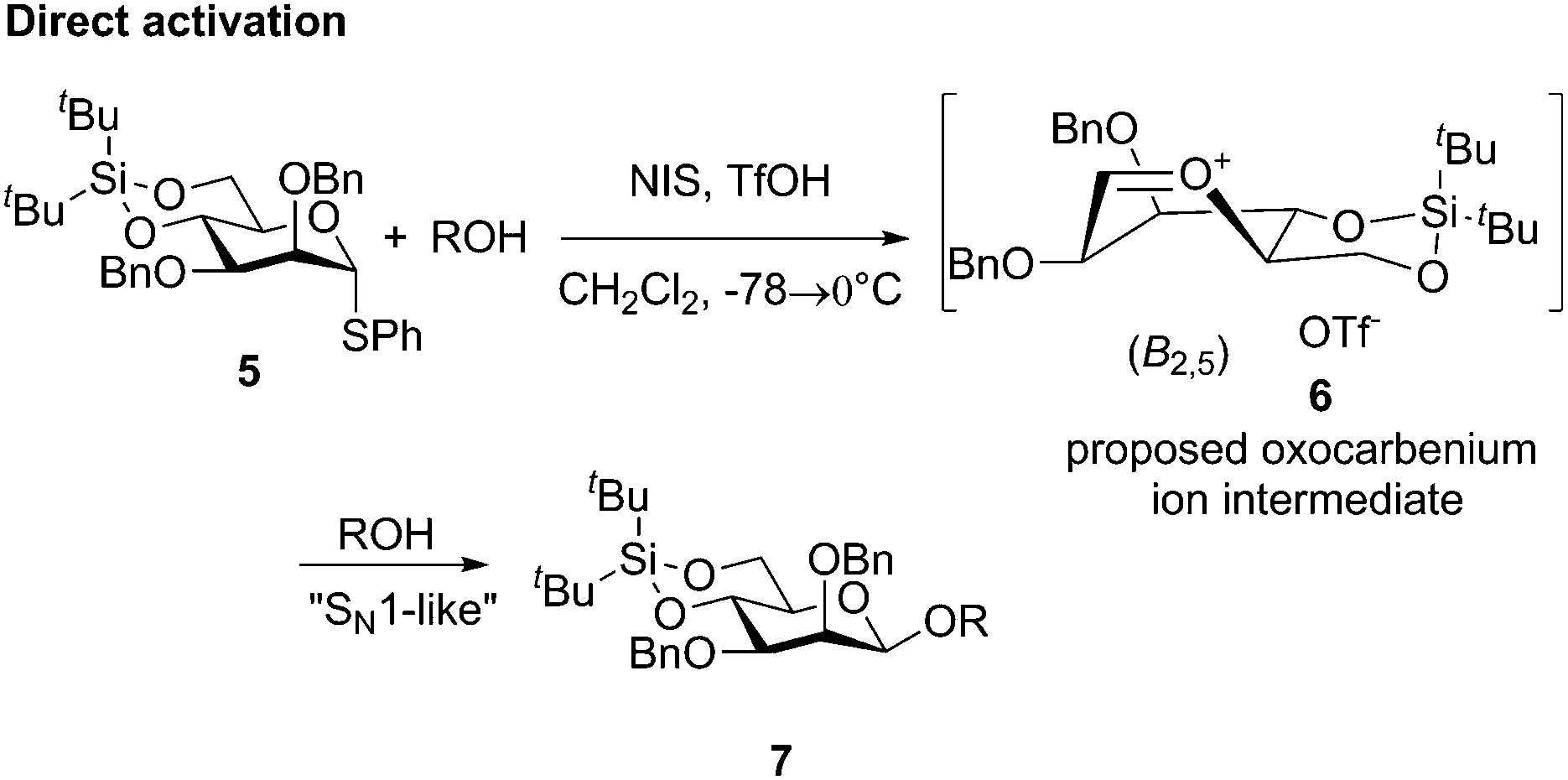 | ||
| Scheme 2 The use of the 4,6-di-tert-butylsilylene tethered donor for selective β-mannosylation using direct activation. | ||
Conclusions made with this 4,6-silylene protected thiomannoside may not hold for the commonly used 4,6-benzylidene protected mannosyl donors. Indeed the literature, including the results reported above, indicated that it did not; NIS-promoted glycosylations with 4,6-benzylidene protected phenyl thiomannosides, as an example, were less β-selective than when using the pre-activation procedure.11 So we were interested in seeing if the apparent indiscriminate β-selectivity of the 4,6-silylene protected thiomannoside 5 was also found for the commonly used benzylidene protected mannosyl donors (such as 1) and if this would also hold for other common donor types such as trichloroacetimidates and sulfoxides. The results of this investigation are reported in the present paper. Benzylidene protected mannosyl donors give β mannosides as the kinetic products and significant α mannoside formation in some cases may be a result of subsequent anomerisation.
As seen above the benzylidene analogue of 5, the α-thioglycoside 8, was reacted with a series of relevant acceptors 11–15 (Scheme 3) promoted by NIS/TfOH. These reactions gave mostly high yield and high β-selectivity (Table 1, entries 1–5).12
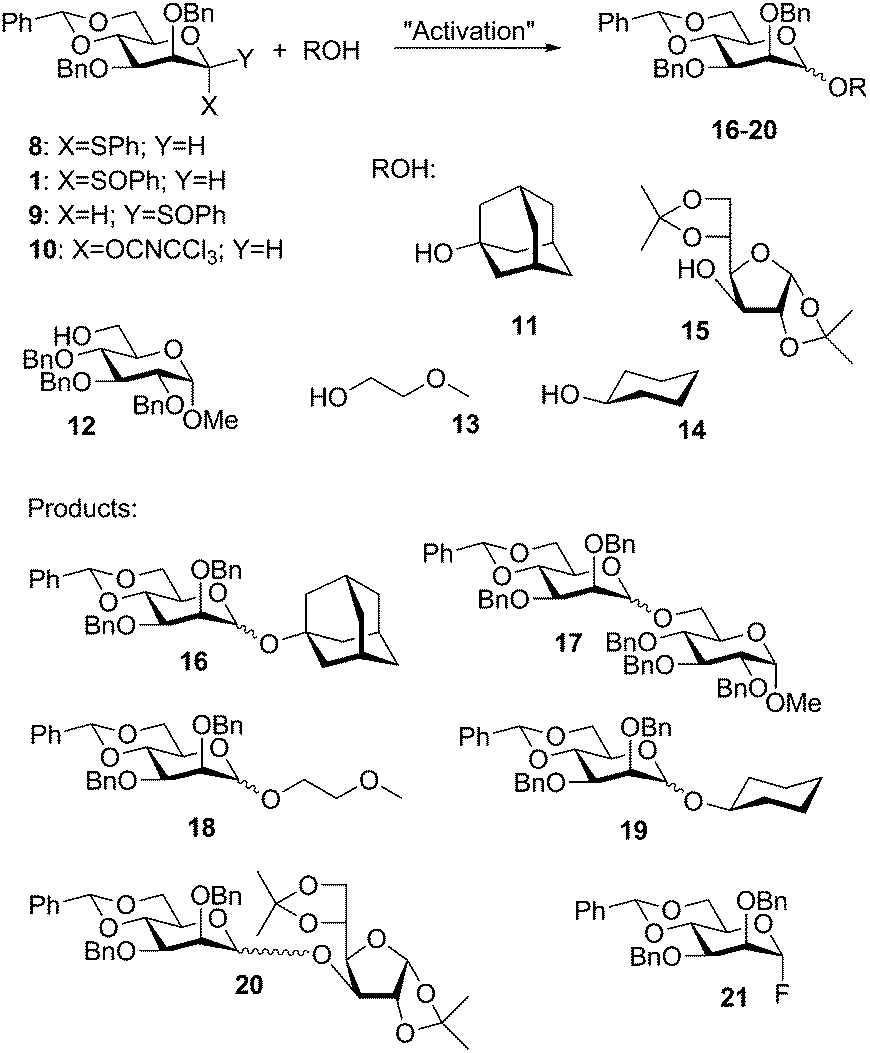 | ||
| Scheme 3 Donors, acceptors and products. | ||
| Entry | Dnr | Ac | Conditions | Yield/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :α :α |
|---|---|---|---|---|
| 0.1 equiv. TfOH was used, ND: not determined, TTBP: 2,4,6-tri-tert-butylpyrimidine, NIS: N-iodosuccinimide and DTBMP = di-tert-butyl-4-methylpyridine. CH2Cl2 is used as solvent in all glycosylations.a Literature reference reaction >9/1 88%.13a,bb 34% 1.c 30% 9.d Preactivation. | ||||
| 1 | 8 | 11 | NIS, TfOH, −78 → 0 °C | 98%/7:1a |
| 2 | 8 | 11 | NIS, TfOH, 0 °C | 94%/6:1 |
| 3 | 8 | 11 | NIS, TfOH, 25 °C | 93%/6:1 |
| 4 | 8 | 12 | NIS, TfOH, 25 °C | 26%/5:1 |
| 5 | 8 | 15 | NIS, TfOH, 25 °C | 34%/2:1b |
| 6 | 1 | 14 | Tf2O, TTBP, −78 °Cd | 82%/>9:1 |
| 7 | 1 | 14 | Tf2O, TTBP, −78 °C | 47%/9:1b |
| 8 | 1 | 15 | Tf2O, DTBMP, −78 °Cd | ND/>20:1 |
| 9 | 1 | 15 | Tf2O, DTBMP, −78 °C | 64%/>20:1 |
| 10 | 1 | 13 | Tf2O, DTBMP, −78 °Cd | 67%/5:1 |
| 11 | 1 | 13 | Tf2O, DTBMP, −78 °C | 42%/5:1 |
| 12 | 9 | 14 | Tf2O, DTBMP, −78 °Cd | ND/β-only |
| 13 | 9 | 14 | Tf2O, DTBMP, −78 °C | 33%/4:1c |
| 14 | 9 | 13 | Tf2O, DTBMP, −78 °C | ND/3:1 |
| 15 | 9 | 14 | Tf2O, DTBMP, −78 °C | 41%/7:2 |
| 16 | 9 | 14 | Tf2O, DTBMP, −78 → rt | 34%/1:12 |
This is essentially the same outcome as with the silylene-tethered donor 510 and comparable to what one obtains with a pre-activation procedure.13 It is noteworthy that β-selectivity is obtained at 0 °C or 25 °C (Table 1, entries 2 & 3) even though α-triflate is not stable at such high temperatures.5c
We now reinvestigated the original protocol for β mannosylation,4,6 which uses α-anomeric sulfoxide 1 that is activated using Kahne's method,14 and included the corresponding β-sulfoxide 9 in the study. When the anomeric set of donors 1 and 9 were preactivated and allowed to react with cyclohexanol 14 at −78 °C both donors gave the β-mannoside 17 with high selectivities (9:1 and β-only respectively – Table 1, entries 6 & 12). When alcohol 14 was present from the beginning, prior to activation, the β:α ratio remained high (9:1 for the α-sulfoxide 1 and 4:1 for the β-sulfoxide 9, Table 1, entries 7 & 13). The acceptor alcohols 13 and 15 gave similar results (Table 1, entries 8–11 & 14). When pre-activation was omitted the yields were significantly lower, which we suspect is due to some triflation of the acceptor alcohol.5c This side reaction becomes a larger problem when the alcohol is a better nucleophile or when the glycosylation is hampered.15 So while preactivation is a good idea as it provides better yields it is not a requirement for β-mannoside formation.
The experiments without pre-activation (Table 1, entries 7, 9, 11 and 13) appears to contradict earlier findings4,6 where such conditions gave either α-mannosides or low selectivity. We speculated that the α-mannoside formation sometimes may be caused by an acid catalyzed anomerisation if insufficient amount of base was present. To test this hypothesis, two reactions were carried out with activation of 9, in the presence of the acceptor 14 (2 equiv.), at −78 °C using sub-stoichiometric amounts of base (1 equiv.) relative to Tf2O (1.7 equiv., Table 1, entries 15 & 16). One reaction was quenched at −70 °C, whereas the other was allowed to reach 0 °C and kept there for 5 min. before quenching it with Et3N. The change in stereoselectivity was dramatic: The first experiment gave mainly β (7:2) and the latter mainly α (1:12). It is clear that, similar to what was observed with the silylene donor,10in situ anomerisation has occurred and obviously led to an erosion of β-selectivity.
We also investigated the popular trichloroacetimidate (TCA) donors. Schmidt has already reported that TCA donor 10 gave β-mannosides promoted by excess TMSOTf,8a but in light of the findings above we wished to see if triflate could be omitted. First we carried out 7 glycosylations with donor 10 and acceptors 11–13 (Table 2, entries 1–7) using TMSOTf catalysis (0.1 equiv.). These experiments confirmed the β-selectivity and yield were high, provided the reaction was run in a nonpolar solvent and quenched at low temperature (entries 1, 3–4) or an acid scavenger (1 equiv. 2,4,6-tri-tert-butylpyrimidine (TTBP))16 was added (entries 2, 5 and 6). If no such precaution against anomerisation was taken the α-anomer was predominant (Table 2, entry 7).
| Entry | Dnr | Ac | Conditions | Yield/β:α |
|---|---|---|---|---|
| Dnr: donor, Ac: acceptor, ND: not determined, NR: no reaction; HFIB: 1,1,1,3,3,3-hexafluoro-2-propanol and TTBP: 2,4,6-tri-tert-butylpyrimidine.a 21 sideproduct.b Quenched after 10% conversion. | ||||
| 1 | 10 | 11 | TMSOTf, CH2Cl2, −78 °C | Quan./>6:1 |
| 2 | 10 | 11 | TMSOTf, TTBP, CH2Cl2, rt | ND/>6:1 |
| 3 | 10 | 11 | TMSOTf, HFIP, −78 °C → rt | 0% |
| 4 | 10 | 11 | TMSOTf, PhMe, −78 °C → rt | 92%/>10:1 |
| 5 | 10 | 12 | TMSOTf, TTBP, CH2Cl2, 25 °C | ND/8:1 |
| 6 | 10 | 13 | TMSOTf, TTBP, CH2Cl2, 0 °C | ND/2.2:1 |
| 7 | 10 | 13 | TMSOTf, CH2Cl2, −78 → rt | ND/1:2.5 |
| 8 | 10 | 11 | BF3·OEt2, CH2Cl2, −78 °C | <5%/>6:1a |
| 9 | 10 | 11 | BF3·OEt2, CH2Cl2, LiOTfb | 10%/>10:1 |
| 10 | 10 | 11 | LiOTf (0.1 equiv.), TTBP, BF3 | ND/>10:1 |
| 11 | 10 | 15 | BF3, CH2Cl2, −78 °C | 21 |
| 12 | 10 | 15 | BF3, CH2Cl2, LiOTf, TTBP | ND/∼5:1 |
| 13 | 10 | 12 | BF3, TTBP, CH2Cl2, 25 °C | 21 |
| 14 | 10 | 12 | TTBP, LiOTf, CH2Cl2, 25 °C | NR |
| 15 | 10 | 12 | TTBP, LiOTf, CH2Cl2, 25 °C, BF3 | ND/>8:1a |
| 16 | 10 | 12 | AgClO4, TTBP, CH2Cl2, 25 °C | NR |
| 17 | 10 | 12 | AgClO4, TTBP, CH2Cl2, 25 °C, BF3 | 21 |
| 18 | 10 | 12 | LiClO4, TTBP, CH2Cl2, 25 °C | NR |
| 19 | 10 | 12 | LiClO4, TTBP, CH2Cl2, 25 °C, BF3 | 21 |
| 20 | 10 | 13 | BF3, CH2Cl2, 25 °C | 77%/1:2a |
To investigate the necessity of triflate in these reactions 13 glycosylations using BF3·OEt2 activation were performed (Table 2, entries 8–20). At low temperature only a small amount of the glycosylation product was obtained, but with β-selectivity (Table 2, entries 8, 11 and 12), while a major sideproduct was mannosyl fluoride 21. At room temperature it was possible to obtain good yield, but now the β-selectivity was lost due to anomerisation (entry 20); adding TTBP did not work here (entry 13). Remarkably, adding LiOTf significantly enhanced the reaction rate. The β-anomer was the kinetic product (entry 9), and adding an acid scavenger allowed full conversion with high β-selectivity (Table 2, entries 10 and 15). LiOTf alone did not activate (entry 14) and silver or lithium perchlorate could not substitute the effect of triflate (entries 16–19).
The above studies show that preactivation is not necessary in order to get good yield and β-selectivity. Regardless of the configuration of the donor the β-mannoside appears to be the kinetic product; however acid catalysed anomerisation, especially at prolonged reaction times at ambient temperature, can erode the stereoselectivity as it gives α-mannoside. This can be prevented by the addition of an acid scavenger.
While triflate is not always necessary in order to get β-selectivity we find it has a clear catalytic effect that appears, at least in some cases, to be caused by the triflate ion itself (Table 2, entry 10). This suggests that formation of a tight ionpair with triflate17 facilitates formation of the oxocarbenium ion and indeed, in accordance with the Crich mechanism,18,19 is an intermediate in the reaction. Recent work with gold-catalyzed glycosylations also indicate that the role of triflate in these reactions is non-trivial.20,21 Perhaps, β-selectivity is attributed to the intermediacy of an oxocarbenium ion ionpair in B2,5 conformation22 either solvent separated as we proposed for the 4,6-silylene donor,10 or as a contact ion pair with triflate in the α-position as recent calculations support.22c This ion pair is glycosylated with β-selectivity either for steric reasons or simply due to the shielding effect of the triflate.
Notes and references
- P. Stallforth, B. Lepenies, A. Adibekian and P. H. Seeberger, J. Med. Chem., 2009, 52, 5561–5577 CrossRef CAS PubMed.
- (a) R. R. Schmidt, Angew. Chem., Int. Ed., 1986, 25, 212–235 CrossRef; (b) S. C. Ranade and A. V. Demchenko, J. Carbohydr. Chem., 2013, 32, 1–43 CrossRef CAS; (c) J. Guo and X.-S. Ye, Molecules, 2010, 15, 7235–7265 CrossRef CAS PubMed; (d) X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed.
- (a) H. Paulsen, Angew. Chem., Int. Ed., 1982, 21, 155–173 CrossRef; (b) K. Toshima and K. Tatsuta, Chem. Rev., 1993, 93, 1503–1531 CrossRef CAS; (c) G. J. Boons, Tetrahedron, 1996, 52, 1095–1121 CrossRef CAS.
- D. Crich and S. Sun, J. Org. Chem., 1996, 61, 4506–4507 CrossRef CAS PubMed.
- The importance of the 4,6-O-benzylidene for β-selectivity was later investigated: (a) D. Crich and A. Banerjee, Org. Lett., 2005, 7, 1395–1398 CrossRef CAS PubMed; (b) H. H. Jensen, L. U. Nordstrøm and M. Bols, J. Am. Chem. Soc., 2004, 126, 9205–9213 CrossRef CAS PubMed; (c) T. G. Frihed, M. T. C. Walvoort, J. D. C. Codée, G. A. van der Marel, M. Bols and C. M. Pedersen, J. Org. Chem., 2013, 78, 2191–2205 CrossRef CAS PubMed.
- D. Crich and S. Sun, Tetrahedron, 1998, 54, 8321–8348 CrossRef CAS.
- (a) D. Crich, Acc. Chem. Res., 2010, 43, 1144–1153 CrossRef CAS PubMed; (b) A. Aubry, K. Sasaki, I. Sharma and D. Crich, Top. Curr. Chem., 2011, 301, 141–188 CrossRef PubMed.
- An SN1-type mechanism has been suggested for C-glycosylations: (a) M. Moumé-Pymbock and D. Crich, J. Org. Chem., 2012, 77, 8905–8912 CrossRef PubMed; (b) M. Huang, P. Retailleau, L. Bohé and D. Crich, J. Am. Chem. Soc., 2012, 134, 14746–14749 CrossRef CAS PubMed.
- For some studies that appear inconsistent with SN2 hypothesis and are precedent to the present work, see: (a) R. Weingart and R. R. Schmidt, Tetrahedron Lett., 2000, 41, 8753–8758 CrossRef CAS; (b) J. D. C. Codée, L. A. Hossain and P. H. Seeberger, Org. Lett., 2005, 7, 3251–3254 CrossRef PubMed; (c) J. Y. Baek, T. J. Choi, H. B. Jeon and K. S. Kim, Angew. Chem., Int. Ed., 2006, 45, 7436–7440 CrossRef CAS PubMed; (d) K. S. Kim, J. H. Kim, Y. J. Lee, Y. J. Lee and J. Park, J. Am. Chem. Soc., 2001, 123, 8477–8481 CrossRef CAS PubMed; (e) T. Tsuda, R. Arihara, S. Sato, M. Koshiba, S. Nakamura and S. Hashimoto, Tetrahedron, 2005, 61, 10719–10733 CrossRef CAS; (f) S. Tanaka, M. Takashina, H. Tokimoto, Y. Fujimoto, K. Tanaka and K. Fukase, Synlett, 2005, 2325–2328 CAS; (g) H. Nagai, S. Matsumura and K. Toshima, Carbohydr. Res., 2003, 338, 1531–1534 CrossRef CAS PubMed; (h) K. Worm-Leonhard, K. Larsen and K. J. Jensen, J. Carbohydr. Chem., 2007, 26, 349–368 CrossRef CAS.
- M. Heuckendorff, J. Bendix, C. M. Pedersen and M. Bols, Org. Lett., 2014, 16, 1116–1119 CrossRef CAS PubMed.
- (a) M. You, Y. Shin, K. H. Chun and J. E. N. Shin, Bull. Korean Chem. Soc., 2000, 21, 562–566 Search PubMed; (b) C. T. Tanifum and C.-W. T. Chang, J. Org. Chem., 2009, 74, 634–644 CrossRef CAS PubMed; (c) S. Wang, D. Lafont, J. Rahkila, B. Picod, R. Leino and S. Vidal, Carbohydr. Res., 2013, 372, 35–46 CrossRef CAS PubMed; (d) D. Crich, M. de la Mora and A. U. Vinod, J. Org. Chem., 2003, 68, 8142–8148 CrossRef CAS PubMed.
- The selectivities are determined from crude NMR obtained after a simple work-up. The spectra corresponding to the α- or β-products are superimposed with the crude spectra to analyze the product distribution. Reference spectra were obtained from purified samples, which were fully assigned and the anomeric stereo-chemistry is determined from the 1JC1,H1-coupling constant and literature references to the known compounds. See K. Bock and C. Pedersen, J. Chem. Soc., Perkin Trans. 2, 1974, 293–297 RSC.
- BSP and Ph2SO are the most common: (a) D. Crich and M. Smith, J. Am. Chem. Soc., 2001, 123, 9015–9020 CrossRef CAS PubMed; (b) J. D. C. Codée, R. E. J. N. Litjens, R. den Heeten, H. S. Overkleeft, J. H. van Boom and G. A. van der Marel, Org. Lett., 2003, 5, 1519–1522 CrossRef PubMed.
- D. Kahne, S. Walker, Y. Cheng and D. B. Engen, J. Am. Chem. Soc., 1989, 111, 6881–6882 CrossRef CAS.
- The method was originally developed for poor acceptors; see ref. 14. It has however also been observed that a less nucleophilic 4-OH acceptor has been triflated (see ref. 5c).
- Addition of TTBP slows down the reaction, but does not inhibit it. As TTBP has a pKa of 1.0 it can potentially be the promotor. For pKa see: H. C. van der Pas and A. Koudijs, Recl. Trav. Chim. Pays-Bas, 1978, 97, 159–161 CrossRef.
- For a discussion of the formation and interconversion of contact and solvent separated ion pairs in glycosylations, see: T. Hosoya, T. Takano, P. Kosma and T. Rosenau, J. Org. Chem., 2014, 79, 7889–7894 CrossRef CAS PubMed.
- M. M. Huang, G. E. Garrett, N. Birlirakis, L. Bohé, D. A. Pratt and D. Crich, Nat. Chem., 2012, 4, 663–667 CrossRef CAS PubMed.
- L. Bohé and D. Crich, Carbohydr. Res., 2015, 403, 48–59 CrossRef PubMed.
- Y. Zhu and B. Yu, Chem. – Eur. J., 2015, 21, 8771–8780 CrossRef CAS PubMed.
- P. Sun, P. Wang, Y. Zhang, X. Zhang, C. Wang, S. Liu, J. Lu and M. Li, J. Org. Chem., 2015, 80, 4164–4175 CrossRef CAS PubMed.
- For studies where a boat conformation has been proposed in β-glycoside formation, see: (a) T. Nukada, A. Bérces and D. M. Whitfield, Carbohydr. Res., 2002, 337, 765–774 CrossRef CAS PubMed; (b) H. Satoh, H. S. Hansen, S. Manabe, W. F. van Gunsteren and P. H. Hünenberger, J. Chem. Theory Comput., 2010, 6, 1783–1797 CrossRef CAS; (c) T. Hosoya, P. Kosma and T. Rosenau, Carbohydr. Res., 2015, 411, 64–69 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures and analysis data. See DOI: 10.1039/c5cc04716a |
| This journal is © The Royal Society of Chemistry 2015 |