
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA ring to rule them all: a cyclic ketene acetal comonomer controls the nitroxide-mediated polymerization of methacrylates and confers tunable degradability†
Vianney
Delplace
a,
Elise
Guégain
a,
Simon
Harrisson
b,
Didier
Gigmes
c,
Yohann
Guillaneuf
c and
Julien
Nicolas
*a
aInstitut Galien Paris-Sud, Université Paris-Sud, UMR CNRS 8612, Faculté de Pharmacie, 5 rue Jean-Baptiste Clément, F-92296 Châtenay-Malabry cedex, France. E-mail: julien.nicolas@u-psud.fr; Web: http://www.twitter.com/julnicolas
bUniversité de Toulouse, CNRS, Laboratoire des Interactions Moléculaires et Réactivité Chimique et Photochimique UMR 5623, 118 route de Narbonne, F-31062 Toulouse, France
cAix-Marseille Université, CNRS, Institut de Chimie Radicalaire UMR 7273, Avenue Escadrille Normandie-Niemen, F-13397 Marseille cedex 20, France
First published on 14th July 2015
Abstract
2-Methylene-4-phenyl-1,3-dioxolane (MPDL) was successfully used as a controlling comonomer in NMP with oligo(ethylene glycol) methyl ether methacrylate (MeOEGMA) to prepare well-defined and degradable PEG-based P(MeOEGMA-co-MPDL) copolymers. The level of ester group incorporation is controlled, leading to reductions in molecular weight of up to 95% on hydrolysis. Neither the polymer nor its degradation products displayed cytoxicity. The method was also successfully applied to methyl methacrylate.
Reversible deactivation radical polymerization (RDRP) enables the synthesis of well-defined, complex and functional macromolecular architectures.1 Nitroxide-mediated polymerization (NMP)1c is perhaps the simplest RDRP technique as it does not require addition of a metal catalyst or radical initiator, but relies on reversible thermal activation of a dormant alkoxyamine end-functionality to form propagating radicals and a stable nitroxide free-radical. Although early drawbacks of NMP (e.g., requirement for high temperatures, applicability to a limited number of monomers), have been substantially overcome by the development of second-generation nitroxides such as SG1,2 the control of methacrylic esters is still a challenge. The high activation-deactivation equilibrium constant (K) of SG1 results in a high concentration of propagating radicals, favoring the occurrence of irreversible termination reactions.3 The polymerization stops at low conversion and highly disperse polymers of uncontrolled molecular weight are obtained. Greatly improved control can be obtained by adding a small amount (typically 2–9 mol%) of a suitable controlling comonomer with favorable kinetic parameters (i.e., low K and low cross-propagation rate constant) such as styrene (S),4 acrylonitrile (AN)5 or 9-(4-vinylbenzyl)-9H-carbazole.6 Most polymer chains were then shown to contain a unit of the controlling comonomer in the terminal position, which explained the high proportion of living chains.4b
The flexibility and robustness offered by RDRP have led to a recent surge in the design of innovative and sophisticated vinyl monomer-based materials intended to find applications in different bio-related areas such as drug delivery or tissue engineering.7 However, because their carbon–carbon backbones resist degradation, these materials may cause prohibitive toxicity, which will hamper their translation to clinical settings and eventually to the market. On this basis, innovative strategies to confer different levels of (bio)degradability to vinyl polymers have emerged. Radical ring-opening polymerization (rROP) of cyclic ketene acetals (CKAs) is one of the most efficient strategies for the incorporation of degradable groups in the polymer backbone, enabling its complete degradation.8 Recent years have seen a resurgence of interest in CKAs, which may be explained by their ability to copolymerize with traditional vinyl monomers by both conventional free-radical polymerization and RDRP techniques. The versatility of this approach has been illustrated by the synthesis of a variety of different copolymer structures,9 which are intended for biomedical applications.
Herein, we report the discovery that 2-methylene-4-phenyl-1,3-dioxolane (MPDL), a scarcely studied CKA,10 both acts as a controlling monomer for the NMP of methacrylic esters and confers tunable degradability to the resulting copolymer, leading to non-cytotoxic degradation products (Scheme 1). This was applied to the SG1-mediated polymerization of methyl methacrylate (MMA) and oligo(ethylene glycol) methyl ether methacrylate (MeOEGMA); the latter being a widely-used monomer for the preparation of non-degradable, PEG-based polymers with potential biological applications.9g,11 The choice of NMP is particularly relevant for the preparation of polymer biomaterials as it has been shown that both SG1-terminated polymers and the SG1 nitroxide itself displayed no cytotoxicity over three different mammalian cell lines even at high doses.5b
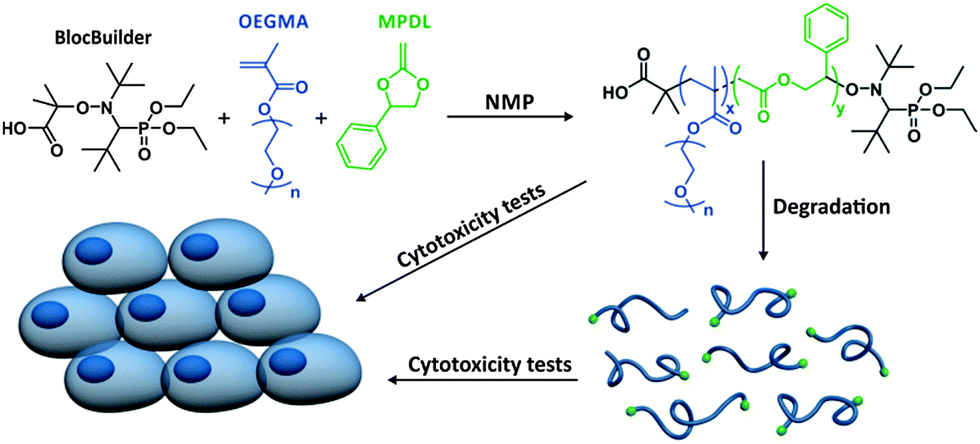 | ||
| Scheme 1 Synthesis, degradation and cytotoxicity of poly[(oligo(ethylene glycol) methyl ether methacrylate)-co-(2-methylene-4-phenyl-1,3-dioxolane)] (P(MeOEGMA-co-MPDL)) prepared by NMP. | ||
MPDL is a 5-membered ring CKA which is easier to synthesize than the more widely used CKAs, 2-methylene-1,3-dioxepane (MDO)12 and 5,6-benzo-2-methylene-1,3-dioxepane (BMDO).13 We recently used MPDL to prepare degradable P(MeOEGMA-co-AN-co-MPDL) terpolymers.9a,14 Analysis by 31P NMR spectroscopy highlighted the competition between AN and MPDL to be the terminal monomer unit, with most copolymer chains terminated by MPDL-SG1 sequences.14 This set us thinking: could MPDL itself act as a controlling comonomer for the NMP of methacrylic esters and at the same time confer tunable degradability to the resulting copolymers?
MPDL was obtained after a multigram-scale synthesis (see ESI† and Fig. S1). The polymerization of MeOEGMA with a variable amount of MPDL (fMPDL,0 = 0–0.7) was initiated by the BlocBuilder alkoxyamine at 90 °C in 50 wt% toluene without additional SG1 (Fig. 1 and Table S1, expts 1–4, ESI†). As expected, the homopolymerization of MeOEGMA (fMPDL,0 = 0) was initially very fast but rapidly stopped, yielding polydisperse PMeOEGMA (Mn = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1, Đ = 1.72) with no control over the molecular weight. When 20 mol% of MPDL was added in the comonomer feed, the situation significantly changed. Two kinetic regimes were observed. The polymerization was rapid within the first 2 h before slowing dramatically, likely due to the insufficient amount of MPDL units inserted to prevent irreversible termination. Some evolution of the Mn with the conversion was however noticed, matching quite well the calculated ones, with substantial improvement over the dispersities. Đ reached 1.54 at 44% conversion for a 13300 g mol−1 P(MeOEGMA-co-MPDL) copolymer. At fMPDL,0 = 0.4, more rapid establishment of first order kinetics with a greater slope over 10 h were observed, presumably due to earlier insertion of MPDL units. This amount of MPDL was perhaps not enough to prevent the occurrence of irreversible termination reactions in the long run, as seen by the deviation from linearity at 24 h. Nevertheless a linear evolution of Mn with monomer conversion between 7000 and 15000 g mol−1, and lower Đ values (i.e., 1.42–1.53) were observed. A further increase in the initial amount of MPDL (fMPDL,0 = 0.7) resulted in immediate establishment of first order kinetics up to high monomer conversion (∼70%) and linear evolution of Mn over a wide range of values (∼4400–27000 g mol−1). Dispersity was also improved as it reached 1.31 at 39% monomer conversion. In fact, the higher fMPDL,0, the lower the dispersities during the polymerization (Fig. 1b). The evolution of molecular weight distribution with time further illustrated the controlled nature of the copolymerizations (Fig. S2, ESI†). Two independent batches of MPDL demonstrated very good reproducibility in terms of kinetics and control, confirming the reliability of this strategy (Fig. S3, ESI†). The copolymers were purified after 30 h by precipitation in cold diethyl ether, giving Mn = 14700 g mol−1 and Đ = 1.55 for expt. 2, Mn = 20100 g mol−1 and Đ = 1.44 for expt. 3, and Mn = 24500 g mol−1 and Đ = 1.30 for expt. 4.
500 g mol−1, Đ = 1.72) with no control over the molecular weight. When 20 mol% of MPDL was added in the comonomer feed, the situation significantly changed. Two kinetic regimes were observed. The polymerization was rapid within the first 2 h before slowing dramatically, likely due to the insufficient amount of MPDL units inserted to prevent irreversible termination. Some evolution of the Mn with the conversion was however noticed, matching quite well the calculated ones, with substantial improvement over the dispersities. Đ reached 1.54 at 44% conversion for a 13300 g mol−1 P(MeOEGMA-co-MPDL) copolymer. At fMPDL,0 = 0.4, more rapid establishment of first order kinetics with a greater slope over 10 h were observed, presumably due to earlier insertion of MPDL units. This amount of MPDL was perhaps not enough to prevent the occurrence of irreversible termination reactions in the long run, as seen by the deviation from linearity at 24 h. Nevertheless a linear evolution of Mn with monomer conversion between 7000 and 15000 g mol−1, and lower Đ values (i.e., 1.42–1.53) were observed. A further increase in the initial amount of MPDL (fMPDL,0 = 0.7) resulted in immediate establishment of first order kinetics up to high monomer conversion (∼70%) and linear evolution of Mn over a wide range of values (∼4400–27000 g mol−1). Dispersity was also improved as it reached 1.31 at 39% monomer conversion. In fact, the higher fMPDL,0, the lower the dispersities during the polymerization (Fig. 1b). The evolution of molecular weight distribution with time further illustrated the controlled nature of the copolymerizations (Fig. S2, ESI†). Two independent batches of MPDL demonstrated very good reproducibility in terms of kinetics and control, confirming the reliability of this strategy (Fig. S3, ESI†). The copolymers were purified after 30 h by precipitation in cold diethyl ether, giving Mn = 14700 g mol−1 and Đ = 1.55 for expt. 2, Mn = 20100 g mol−1 and Đ = 1.44 for expt. 3, and Mn = 24500 g mol−1 and Đ = 1.30 for expt. 4.
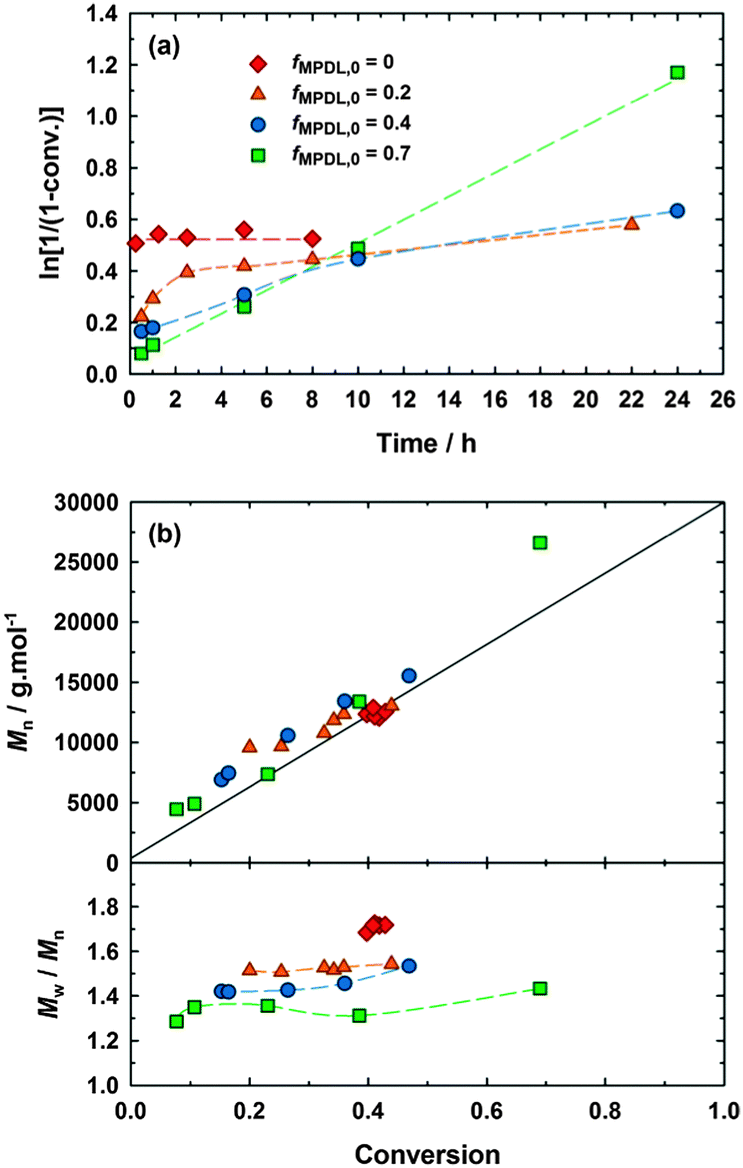 | ||
| Fig. 1 NMP of MeOEGMA and MPDL in toluene initiated by the BlocBuilder alkoxyamine at 90 °C, as a function of the initial amount of MPDL: ◆, expt. 1 (fMPDL,0 = 0); ▲, expt. 2 (fMPDL,0 = 0.2); ●, expt. 3 (fMPDL,0 = 0.4); ■, expt. 4 (fMPDL,0 = 0.7). (a) Ln[1/(1 − conv.)] vs. time (conv. = MeOEGMA conversion). (b) Number-average molar mass, Mn, and dispersity, Mw/Mn, vs. conversion. The full line represents the theoretical Mn (MeOEGMA) and the dashed ones are guides for the eye only. | ||
1H NMR spectroscopy of the purified P(MeOEGMA-co-MPDL) copolymers (expts 2–4) showed all signals expected for a P(MeOEGMA-co-MPDL) structure (Fig. S4, ESI†). The aromatic protons of the phenyl group of MPDL (signal e) were clearly visible and confirmed that its level of incorporation in the copolymer could be adjusted by varying its initial concentration in the comonomer feed. The preferential formation of the MPDL-SG1 terminal sequence was confirmed by 31P NMR spectroscopy as its spectrum is very similar to that of PS-SG1 (Table S1, expt. 5, ESI†), due to the styrene-like ring-opened structure of MPDL (Fig. S5, ESI†). This analogy in structures between the open radical forms of S and MPDL explains the ability of MPDL to act as a controlling comonomer. The copolymerization was also successfully performed with MMA, a representative methacrylate monomer (Table S1, expt. 6, ESI†). Under similar experimental conditions, a well-defined P(MMA-co-MPDL) copolymer (conv. MMA (15 h) = 40%, Mn = 10200 g mol−1, Đ = 1.20) was prepared (Fig. S6, ESI†). 31P NMR spectroscopy performed in the presence of a known amount of diethyl phosphite as internal reference gave a living chain fraction as high as 85% (Fig. S7, ESI†), which is similar to those obtained using S or AN as a comonomer, thus establishing the efficiency of MPDL as a controlling comonomer.
The molar fraction of MPDL in the copolymer, FMPDL, with respect to MeOEGMA was determined by 1H NMR spectroscopy to be 3.6, 11.3 and 24.8 mol% for expt. 2 (fMPDL,0 = 0.2), expt. 3 (fMPDL,0 = 0.4) and expt. 4 (fMPDL,0 = 0.7), respectively. As for the vast majority of CKAs, unfavorable reactivity ratios impose high concentrations of MPDL in the comonomer feed in order to achieve substantial incorporation in the resulting copolymer. The reactivity ratios were determined by fitting the integrated form of the copolymer composition equation to the monomer feed composition vs. conversion data using the visualization of the sum of squares method,15 assuming non-negligible errors in both variables. This procedure gave a point estimate of 6.95 for rMeOEGMA and 0 for rMPDL, with a 95% joint confidence region spanning the range of 6.3–7.9 for rMeOEGMA and 0–0.038 for rMPDL (Fig. S8, ESI†). The values obtained are typical for copolymerizations of methacrylate monomers with ring-opening monomers such as MDO (rMMA = 34, rMDO = 0.057),16 1,1-dichloro-2-vinylcyclopropane (rMMA = 11, rVCP = 0.07)17 and 7-methylene-2-methyl-1,5-dithiacyclooctane (rMMA = 6.3, rMDTO = 0.52),18 and reflect the lack of radical-stabilizing substituents on the double bond of the CKA.
To assess the degradability, hydrolytic degradation of the copolymers was performed in accelerated conditions (i.e., 5% KOH aqueous solution) and monitored over a period of 24 h to estimate the kinetics of hydrolysis (Fig. 2a). Adjustable incorporation of MPDL in the copolymer enabled fine tuning of the level of degradation, which typically reached its steady state after 5 h. While the copolymer with the lowest amount of MPDL (expt. 2) led to modest degradation (∼30% decrease in Mn for FMPDL = 0.036), significant (even complete) degradation was observed for greater amounts of MPDL. When FMPDL = 0.113 (expt. 3), the decrease in Mn was 81% whereas it reached 95% for FMPDL = 0.248 (expt. 4). The complete hydrolysis of the latter was confirmed by size exclusion chromatograms which shifted toward lower molar masses (Fig. 2b). Note that the polymer without MPDL (expt. 1) was perfectly stable, thus ruling out reduction in molecular weight due to ester side chain hydrolysis.
 | ||
| Fig. 2 Hydrolytic degradation in 5% KOH of P(MeOEGMA-co-MPDL) as a function of the content in MPDL: ◆, expt. 1 (FMPDL,0 = 0); ▲, expt. 2 (FMPDL,0 = 0.036); ●, expt. 3 (FMPDL,0 = 0.113); ■, expt. 4 (FMPDL,0 = 0.248). (a) Evolution of the number-average molar mass, Mn, with time. Dashed lines are guides for the eye only. (b) Evolution of the SEC chromatograms at different time for expt. 4 during degradation. | ||
The theoretical decrease in Mn of these copolymers can be calculated from the average sequence length of PMeOEGMA blocks in each copolymer. This can be directly estimated from the copolymer composition, as due to the very low rMPDL, the probability of finding a sequence of two or more MPDL units is negligible. From the MPDL content of the polymers it corresponds to, on average, 1 MPDL unit for every 26.8 (expt. 2), 7.8 (expt. 3) and 3.0 (expt. 4) MeOEGMA units. These lengths correspond to Mn of 8210, 2530 and 1080 g mol−1, respectively. These values are in rather good agreement with those obtained for the degraded polymers (12600, 3500 and 800 g mol−1, respectively), especially that with the highest amount of MPDL, thus confirming the near-complete hydrolysis of the main–chain ester groups.
Conferring PEG-based polymers with the ability to be degraded in a hydrolytic environment is a significant advance compared to traditional linear or comb-like PEGs that are resistant to degradation. However, neither the degradation products nor the polymer itself should be cytotoxic in an optimized product. This point is crucial as it will determine the ultimate biocompatibility of the materials. The potential toxicity of different copolymers has been tested on two representative mammalian cell types: murine fibroblasts (NIH/3T3) and murine macrophages (J774.A1). Whereas NIH/3T3 cells are one of the most commonly used fibroblast cell lines, J774.A1 cells, which play a key role in phagocytosis, were chosen to highlight possible toxicity of the copolymers after being engulfed by macrophages. The cytotoxicity was evaluated in vitro by MTT assay at two copolymer concentrations (0.1 and 1 mg mL−1) (Fig. 3). Incubation of undegraded P(MeOEGMA-co-MPDL) copolymer (expt. 3, FMPDL,0 = 0.113) with both cell lines resulted in >95% cell viability at 0.1 mg mL−1. At 1 mg mL−1 cell viabilities were still high; 83% with NIH/3T3 cells and 72% with J774.A1 cells. More importantly, MTT assays revealed no cytotoxicity from the degradation products whatever the cell line and the copolymer concentration. Cell viabilities were in the 97–99% range at 0.1 mg mL−1 and 86–92% at 1 mg mL−1. These results indicate the absence of obvious toxicity from the copolymer and from the products, which is a very encouraging indication of their biocompatibility and their safe use as biomaterial building blocks.
 | ||
| Fig. 3 Cell viability (MTT assay) after incubation of NIH/3T3 cells and J774.A1 cells with P(MeOEGMA-co-MPDL) copolymer (expt. 3, FMPDL,0 = 0.113) at 0.1 and 1 mg mL−1. Results were expressed as percentages of absorption of treated cells (±SD) in comparison to that of untreated ones as a control. | ||
In conclusion, we have demonstrated that MPDL, a little-studied CKA, can both act as a controlling comonomer for the SG1-mediated polymerization of methacrylic esters and confer tunable degradability to the resulting copolymer. By adjusting the comonomer feed, up to complete degradation was observed. No significant toxicity was shown, either from P(MeOEGMA-co-MPDL) or from its degradation products. Not only does this broaden the range of suitable controlling comonomers for the NMP of methacrylic esters, but it adds significant value to the copolymerization approach due to the degradability of the resulting copolymer. Additionally, this study opens up exciting perspectives in the design of new controlling comonomers and NMP-derived degradable architectures with a wide range of potential (bio)applications. For instance, nanoparticles or therapeutic proteins decorated with degradable PEG segments should show reduced toxicity and persistence compared to their nondegradable PEGylated analogues.
The manuscript was written through contributions of all authors. The authors thank the French National Research Agency (ANR-11-JS08-0005) for the financial support of the PhD thesis of VD and the French Ministry of Research for the financial support of the PhD thesis of EG. Arkema is warmly acknowledged for kindly providing the BlocBuilder MA alkoxyamine and the SG1 nitroxide. CNRS is also acknowledged for financial support.
Notes and references
- (a) M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS PubMed; (b) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402 CrossRef CAS; (c) J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63 CrossRef CAS PubMed; (d) S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347 CrossRef CAS PubMed.
- (a) D. Benoit, V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc., 1999, 121, 3904 CrossRef CAS; (b) D. Benoit, S. Grimaldi, S. Robin, J.-P. Finet, P. Tordo and Y. Gnanou, J. Am. Chem. Soc., 2000, 122, 5929 CrossRef CAS.
- C. Dire, J. Belleney, J. Nicolas, D. Bertin, S. Magnet and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6333 CrossRef CAS PubMed.
- (a) B. Charleux, J. Nicolas and O. Guerret, Macromolecules, 2005, 38, 5485 CrossRef CAS; (b) J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. A. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39, 8274 CrossRef CAS.
- (a) J. Nicolas, S. Brusseau and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 34 CrossRef CAS PubMed; (b) M. Chenal, S. Mura, C. Marchal, D. Gigmes, B. Charleux, E. Fattal, P. Couvreur and J. Nicolas, Macromolecules, 2010, 43, 9291 CrossRef CAS.
- (a) B. Lessard, E. J. Y. Ling, M. S. T. Morin and M. Marić, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1033 CrossRef CAS PubMed; (b) B. H. Lessard, E. J. Y. Ling and M. Marić, Macromolecules, 2012, 45, 1879 CrossRef CAS; (c) B. H. Lessard, Y. Guillaneuf, M. Mathew, K. Liang, J.-L. Clement, D. Gigmes, R. A. Hutchinson and M. Marić, Macromolecules, 2013, 46, 805 CrossRef CAS.
- (a) V. Delplace, P. Couvreur and J. Nicolas, Polym. Chem., 2014, 5, 1529 RSC; (b) M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545 RSC.
- S. Agarwal, Polym. Chem., 2010, 1, 953 RSC.
- (a) V. Delplace, A. Tardy, S. Harrisson, S. Mura, D. Gigmes, Y. Guillaneuf and J. Nicolas, Biomacromolecules, 2013, 14, 2837 CrossRef PubMed; (b) J.-F. Lutz, J. Andrieu, S. Üzgün, C. Rudolph and S. Agarwal, Macromolecules, 2007, 40, 8540 CrossRef CAS; (c) J. Undin, A. Finne-Wistrand and A.-C. Albertsson, Biomacromolecules, 2013, 14, 2095 CrossRef CAS PubMed; (d) J. Undin, T. Illanes, A. Finne-Wistrand and A.-C. Albertsson, Polym. Chem., 2012, 3, 1260 RSC; (e) S. Maji, M. Zheng and S. Agarwal, Macromol. Chem. Phys., 2011, 212, 2573 CrossRef CAS PubMed; (f) Y. Zhang, D. Chu, M. Zheng, T. Kissel and S. Agarwal, Polym. Chem., 2012, 3, 2752 RSC; (g) C. Riachi, N. Schüwer and H.-A. Klok, Macromolecules, 2009, 42, 8076 CrossRef CAS; (h) I. S. Chung and K. Matyjaszewski, Macromolecules, 2003, 36, 2995 CrossRef CAS; (i) D. J. Siegwart, S. A. Bencherif, A. Srinivasan, J. O. Hollinger and K. Matyjaszewski, J. Biomed. Mater. Res., Part A, 2008, 87, 345 CrossRef PubMed; (j) Y. Zhang, A. Aigner and S. Agarwal, Macromol. Biosci., 2013, 13, 1267 CrossRef CAS PubMed.
- W. J. Bailey, S. R. Wu and Z. Ni, Makromol. Chem., 1982, 183, 1913 CrossRef CAS PubMed.
- (a) B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563 RSC; (b) J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459 CrossRef CAS PubMed; (c) J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083 CrossRef CAS PubMed.
- W. J. Bailey, Z. Ni and S. R. Wu, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 3021 CrossRef CAS PubMed.
- W. J. Bailey, Z. Ni and S. R. Wu, Macromolecules, 1982, 15, 711 CrossRef CAS.
- V. Delplace, S. Harrisson, A. Tardy, D. Gigmes, Y. Guillaneuf and J. Nicolas, Macromol. Rapid Commun., 2014, 35, 484 CrossRef CAS PubMed.
- M. Van Den Brink, A. M. Van Herk and A. L. German, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3793 CrossRef CAS.
- G. E. Roberts, M. L. Coote, J. P. A. Heuts, L. M. Morris and T. P. Davis, Macromolecules, 1999, 32, 1332 CrossRef CAS.
- T. Takahashi, J. Polym. Sci., Part A: Polym. Chem., 1970, 8, 739 CrossRef CAS PubMed.
- S. Harrisson, T. P. Davis, R. A. Evans and E. Rizzardo, Macromolecules, 2001, 34, 3869 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, SEC chromatograms, additional kinetics, 1H and 31P NMR spectra, determination of the reactivity ratios. See DOI: 10.1039/c5cc04610f |
| This journal is © The Royal Society of Chemistry 2015 |