
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic CO2 reduction using visible light by metal-monocatecholato species in a metal–organic framework†
Yeob
Lee
a,
Sangjun
Kim
b,
Honghan
Fei
a,
Jeung Ku
Kang
*b and
Seth M.
Cohen
*a
aDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, USA. E-mail: scohen@ucsd.edu
bGraduate School of EEWS, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 305-701, Republic of Korea
First published on 24th September 2015
Abstract
Metal–organic frameworks (MOFs) with isolated metal-monocatecholato groups have been synthesized via postsynthetic exchange (PSE) for CO2 reduction photocatalyst under visible light irradiation in the presence of 1-benzyl-1,4-dihydronicotinamide and triethanolamine. The Cr-monocatecholato species are more efficient than the Ga-monocatecholato species.
The conversion of CO2 into hydrocarbons has attracted great attention owing to global warming caused, in part, by CO2 from fossil fuel combustion.1,2 Inspired by photosynthesis, development of an artificial system that catalytically regenerates hydrocarbon fuels from CO2, H2O, and sunlight is one very intriguing approach.3–5 Artificial photosynthesis would consist of two reactions: water oxidation to extract electrons from water and CO2 reduction to generate carbonaceous radicals using electrons generated from water oxidation. CO2 requires a large driving force to be transformed to other compounds due to the high kinetic and thermodynamic stability of CO2.6–8 Several photocatalytic systems for CO2 reduction, including heterogeneous semiconductor systems and homogeneous transition metal-based complexes have been investigated but challenges remain. For example, many metal oxides are active under only UV light, which represents only ∼4% of the solar energy spectrum. This has encouraged research on extending the light absorption edge of metal oxides.9 Homogeneous metal complexes based on Ru, Re, and Ir have been investigated; however, an inability to recycle and reuse these precious metal compounds remains a limitation of these systems.10
Metal–organic frameworks (MOFs) are hybrid materials that consist of secondary building units (SBUs) and organic linkers. The rational design of MOFs with tunable properties through a selective combination of metal ions and organic ligands has produced materials useful for various applications. Photocatalytic applications of MOFs also have been studied.11–15 Lin and co-workers reported a MOF photocatalyst doped with Re(bpy)(CO)3Cl complexes that reduced CO2 to CO under UV light irradiation.11 This pioneering work for MOF photocatalysts showed poor efficiency due, in part, to a low doping of the Re catalyst into the MOF. Fu et al. synthesized visible-light sensitive NH2-MIL-125(Ti) with an amine-functionalized organic linker. This material reduced CO2 to HCOO− in the presence of triethanolamine (TEOA) under visible-light irradiation.12 In addition, Li et al. developed a non-porous coordination polymer consisting of Y metal ions and Ir(ppy)2(dcbpy) metalloligands; this material reduced CO2 to HCOO− under visible light irradiation.16 Despite these advances, the development of MOF photocatalysts for CO2 reduction is still in its infancy.
Herein, we report a new MOF photocatalysts that incorporate catalytic metal sites, using postsynthetic modification methods, for CO2 reduction to formic acid in the presence of 1-benzyl-1,4-dihydronicotinamide (BNAH) and triethanolamine (TEOA). The Zr(IV)-based MOF (UiO-66, UiO = University of Oslo) was subjected to postsynthetic exchange (PSE)17–20 with a catechol-functionalized organic linker (catbdc, 2,3-dihydroxyterephthalic acid, to produce UiO-66-CAT).19 Two different trivalent metal ions, Cr(III) and Ga(III), were then incorporated into the catbdc sites to afford unprecedented Cr- and Ga-monocatecholato species in a robust UiO-66. The catbdc organic linkers are responsible for visible light absorption and metalation by Cr(III) and Ga(III) facilitates electron transfer within the MOFs.
PSE has become a facile and efficient strategy to functionalize MOFs under mild conditions (Fig. 1). UiO-66 was prepared solvothermally in DMF containing 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio mixture of ZrCl4 and H2bdc with acetic acid as a modulator at 120 °C. UiO-66 was then exposed to DMF/H2O solution containing 2 equiv. catbdc at 85 °C to achieve PSE into UiO-66.19 This gave a UiO-66 derivative that contained ∼34% catbdc and ∼66% bdc ligand. The metalation of the catechol functionality in UiO-66-CAT was conducted using aqueous K2CrO4 under acidic conditions (pH = 3). After incubation at room temperature (1 h) the pale yellow UiO-66-CAT changed to a dark brown color. Similarly, an aqueous solution of Ga(NO3)3(H2O)x was used to achieve metalation with Ga(III). After metalation, the MOFs were isolated by centrifugation, washed extensively with deionized water and MeOH, and dried under vacuum. Inductively coupled plasma mass spectrometry (ICP-MS, Table S1, ESI†) confirmed the atomic ratio of 0.25 (Cr/Zr) and 0.26 (Ga/Zr), indicating that ∼80% of the available catbdc ligands were metalated by both the Cr and Ga procedures based on three independent samples.
:1 molar ratio mixture of ZrCl4 and H2bdc with acetic acid as a modulator at 120 °C. UiO-66 was then exposed to DMF/H2O solution containing 2 equiv. catbdc at 85 °C to achieve PSE into UiO-66.19 This gave a UiO-66 derivative that contained ∼34% catbdc and ∼66% bdc ligand. The metalation of the catechol functionality in UiO-66-CAT was conducted using aqueous K2CrO4 under acidic conditions (pH = 3). After incubation at room temperature (1 h) the pale yellow UiO-66-CAT changed to a dark brown color. Similarly, an aqueous solution of Ga(NO3)3(H2O)x was used to achieve metalation with Ga(III). After metalation, the MOFs were isolated by centrifugation, washed extensively with deionized water and MeOH, and dried under vacuum. Inductively coupled plasma mass spectrometry (ICP-MS, Table S1, ESI†) confirmed the atomic ratio of 0.25 (Cr/Zr) and 0.26 (Ga/Zr), indicating that ∼80% of the available catbdc ligands were metalated by both the Cr and Ga procedures based on three independent samples.
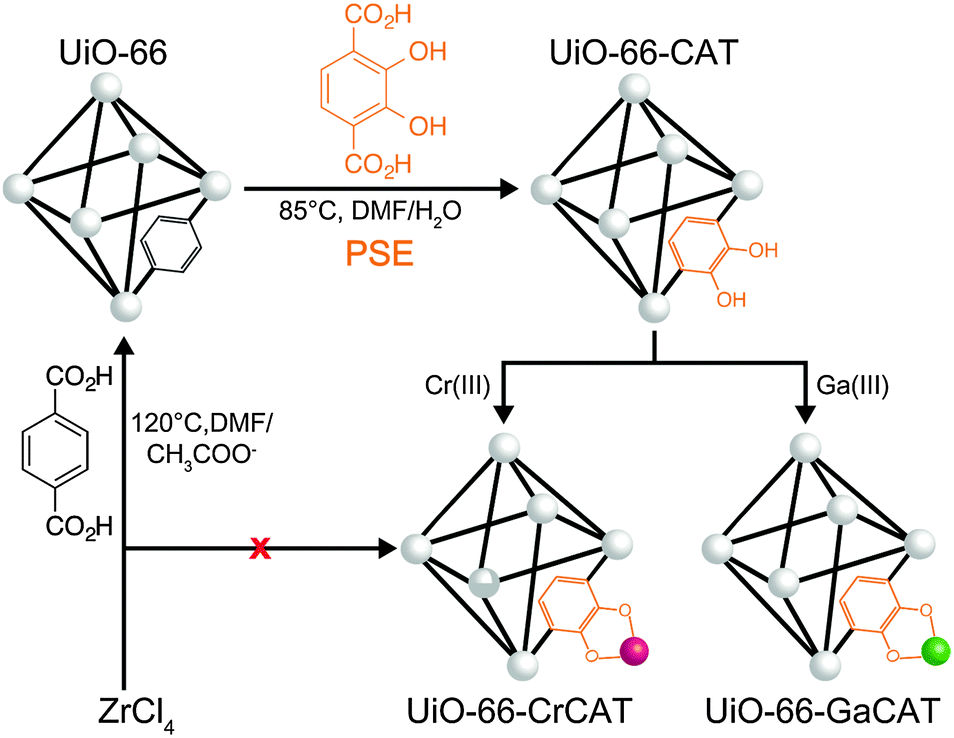 | ||
| Fig. 1 Preparation of MOF photocatalysts through postsynthetic exchange (PSE) and metalation. | ||
The MOFs formed as nanocrystallites ∼150 nm on an edge, with an octahedral morphology. The crystallinity of the MOFs did not change upon PSE or metalation as evidenced by the PXRD patterns as shown in Fig. 2a. Scanning electron microscopy (SEM) images showed that the morphology and crystal size of UiO-66-CrCAT and UiO-66-GaCAT were also unchanged from the parent UiO-66 material (Fig. S1 and S2, ESI†). The characteristic M(III) signals was detected using energy dispersive X-ray spectroscopy (Fig. S1, ESI†).
 | ||
| Fig. 2 (a) PXRD patterns of UiO-66 and functionalized UiO-66 derivatives; (b) UV-vis spectroscopy for UiO-66, UiO-66CAT, UiO-66-CrCAT, and UiO-66-GaCAT. F(R) were calculated from diffuse reflectance measurement; X-ray photoelectron spectroscopy analysis of (c) UiO-66-CrCAT and (d) UiO-66-GaCAT. | ||
The UV-visible spectroscopy of the MOFs was altered upon PSE and metalation as illustrated in Fig. 2b. The diffuse reflectance of samples was measured and the reflectance values were subjected to the Kubelka–Munk function ((1 − R)2/2R) to quantify the light absorption ability of samples. H2bdc derivatives with electron donating functionality such as NH2, OH, and SH are known to increase the HOMO level of H2bdc.21–23 Thus, UiO-66-CAT is expected to absorb some visible light as a result of the catechol groups. A color change to dark brown was observed upon metalation with Cr(III). As expected, Cr(III) binding to catbdc results in the generation of ligand-to-metal charge transfer (LMCT).24,25 A similar color change was not observed upon metalation with Ga(III), as expected for this closed-shell ion.
X-ray photoelectron spectroscopy (XPS) of UiO-66-CrCAT and UiO-66-GaCAT was carried out to determine the oxidation states of Cr and Ga. The 2p orbital information was obtained and each spectrum exhibits two peak contributions. Chromium oxide (Cr2O3) and gallium oxide (Ga2O3) were selected as references for both UiO-66-M(III)CATs. UiO-66-CrCAT shows two peaks at 586.10 eV and 576.58 eV corresponding to 2p1/2 and 2p3/2 binding energies, respectively. These values compare well with energy levels in Cr2O3 (586.13 eV and 576.33 eV). This indicates that the Cr(VI) in K2CrO4 was reduced to Cr(III) upon metalation, as previously observed.19 The XPS for UiO-66-GaCAT also consists of two peaks at 1144.79 eV and 1117.89 eV corresponding to 2p1/2 and 2p3/2 energy levels, respectively. These values match well to binding energies in Ga2O3 (1144.67 eV and 1117.79 eV). Therefore, XPS of UiO-66-CrCAT and UiO-66-GaCAT confirm the trivalent oxidation state of Cr and Ga in these MOFs.26,27
These M(III)-monocatecholato functionalized MOFs were investigated for their photocatalytic CO2 reduction activity. The MOFs were introduced into a mixed solution of 4:1 (v/v) MeCN and TEOA, which contained BNAH (0.1 M). In this photocatalytic reaction, BNAH serves as a reductant for CO2 to produce carbonaceous radicals28,29 and TEOA acts as a sacrificial base to capture protons from BNAH.30 The product solutions were found to consist of water, ethyl acetate, MeCN, and HCOOH. The photocatalytic activity of each UiO-66-M(III)CAT are shown in Fig. 3a. Turnover numbers were calculated from the amount of HCOOH produced versus the number of M(III)-catecholato sites in each MOF. Turnover numbers were calculated as 11.22 ± 0.37 for UiO-66-CrCAT and 6.14 ± 0.22 for UiO-66-GaCAT, respectively. UiO-66-CrCAT and UiO-66-GaCAT produced 51.73 ± 2.64 μmoles and 28.78 ± 2.52 μmoles of HCOOH from CO2 photocatalysis, respectively (6 h of visible light irradiation). These numbers indicate that each Cr-catecholato species catalyzed the conversion of ∼11 CO2 molecules, while each Ga-catecholato species catalyzed the conversion of ∼6 CO2 molecules over the 6 h reaction time. UiO-66-CrCAT proved to be a more efficient catalyst than UiO-66-GaCAT under these reaction conditions. Both UiO-66-M(III)CATs produced negligible amount of H2 and CO that can be generated from photocatalysis of CO2 in the presence of TEOA and BNAH. This indicates that metalated catecholato species are suitably selective for CO2 reduction to formate.
 | ||
| Fig. 3 (a) Photocatalytic ability of UiO-66-CrCAT (red) and UiO-66-GaCAT (green) over three cycles (6 h per cycle); (b) 13C liquid NMR spectra of photocatalytic CO2 reduction by UiO-66-CrCAT (red) and UiO-66-GaCAT (green) after 13 h. | ||
Both MOFs were tested over three catalytic cycles to investigate the stability and reusability of the MOF photocatalysts. Samples were recovered by centrifugation, washed with copious amounts of MeOH, and activated under vacuum after each cycle. The numbers of M(III)-catecholato sites were also redetermined for each cycle to obtain accurate turnover numbers. The catalytic activities of both MOFs were relatively unchanged over the three cycles (Fig. 3a and Fig. S4, ESI†). However, a small amount of M(III) ions leached from the MOFs based on an decreasing M(III)/Zr(IV) ratio as determined by ICP-MS (Table S1, ESI†) after each reaction. The photocatalysis results in Fig. 3a were reproducible based on findings from three independent samples (Fig. S5, ESI†). Quantum yields for both UiO-66-M(III)CAT MOFs were obtained under monochromatic light irradiation using a band-pass optical filter (450 nm). UiO-66-CrCAT showed a higher quantum yield value (1.83 ± 0.16%) when compared to UiO-66-GaCAT (1.17 ± 0.11%). The Fe(III) metalated UiO-66-CAT was also prepared following a previous report.19 Under identical photocatalytic conditions, UiO-66-FeCAT (Fe:Zr = 0.27, ∼80% of catbdc metalated) produced little HCOOH (1.47 μmoles, Table S2, ESI†). The redox potential for Fe(III) is not suitable (0.77 V vs. SHE) for CO2 reduction (unlike Cr(III) and Ga(III)), and hence this MOF derivative is not a suitable photocatalyst.
The use of UiO-66-CAT prior to metalation as a photocatalyst did not produce HCOOH as measured by GC-MS (and 13C NMR, see below). This rules out the catbdc ligand alone or the Zr6 SBU clusters as the catalytic sites for reduction. UiO-66-CAT is not suited to accept photo-generated electrons from the catbdc ligand because the redox potential of Zr6 SBU is higher than the LUMO of bdc linkers.15,31 The control reactions with UiO-66-CAT support that photocatalytic reduction of CO2 is dependent on the M(III)-catecholato species in the MOF.
13C NMR spectra was acquired using a 13C isotope of CO2 as a substrate, in order to confirm the origin of the carbon source for HCOOH upon photocatalysis with MOFs. The photocatalytic conditions used were identical to those described above except for the use of CD3CN and with the reactor purged by 13CO2 (∼99% of 13C) before light irradiation. After 13 h, the reaction mixture was transferred to an NMR tube without further treatment (i.e. no H2SO4). Reference solutions were prepared for H13COOH and the results are shown in Fig. S5 (ESI†). The chemical shift for H13COOH in CD3CN was found at 162.73 ppm; however, this value was increased to 169.86 ppm in 4:1 CD3CN:TEOA solution due to deprotonation to H13COO−.30,32 In addition, it was expected that 13CO2 could form 13CO32− and H13CO3− under the alkaline reaction conditions.33
Several signals corresponding to H13COO−, 13CO2, 13CO32−, H13CO3−, MeCN, and other small signals were found in 13C NMR spectrum of product solution from both UiO-66-M(III)CAT reaction mixtures (Fig. 3b). Small signals were also observed from BNAH isotopes and these signals were also found in reference solution of CD3CN:TEOA with BNAH. Moreover, using unlabeled 12CO2, instead of 13CO2, produced spectra with no H13COO− peak (Fig. S6, ESI†). The strong resonance peaks observed for H13COO− indicate that the carbon source is primarily from CO2 gas, not from decomposition of the MOFs.
MOFs containing M(III)-monocatecholato species after three cycles of photocatalysis were characterized using PXRD and SEM measurements to evaluate their structural and chemical integrity, which is another essential characteristic feature for an optimal photocatalyst. Both UiO-66-CrCAT and UiO-66-GaCAT maintained their crystalline structure over 18 h of exposure to the photocatalysis conditions (although some Cr and Ga leaching was observed, Table S1, ESI†). Crystal size, morphology, and surface texture (Fig. S7, ESI†) remained intact throughout photocatalysis, indicating the SBUs and organic linkers were not significantly degraded. Furthermore, the crystallinity was maintained in both UiO-66 derivatives during 1 week of light irradiation (Fig. S8, ESI†). Lastly, XPS results show that both Cr and Ga remained in their trivalent oxidation states after photocatalysis (Fig. S9, ESI†). The BET specific surface area of both UiO-66-CrCAT and UiO-66-GaCAT was slightly decreased after three cycles of photocatalysis, which indicates that some pores were blocked or collapse occured during intense photoirradiation; however, the surface areas indicated that the MOFs were still highly porous (Fig. S10, ESI†). The data suggest that isolated M(III)-monocatecholato functionalities residing in MOFs acted as independent photocatalytic sites.
Photoluminescence (PL) spectroscopy was carried out to characterize the charge transfer between catbdc and M(III) in UiO-66-M(III)CAT. As shown in Fig. 4a, the emission intensity of UiO-66-CAT is significantly reduced after metalation. This indicates that the recombination rate of photogenerated electron–hole pairs in the organic linkers was markedly decreased upon metalation, suggesting that charges were transferred to other sites in the MOFs. The decreases in emission intensity were different for UiO-66-CrCAT and UiO-66-GaCAT, suggesting that the metalated catbdc centers are the acceptors. This indicates that LMCT occurred between the catechol units and metal elements, as expected.34 In particular, UiO-66-CrCAT quenched ∼80% of the photo-generated charges in catbdc and this value is close to the ratio of Cr-bound catechol ligands in UiO-66-CrCAT. This increased electron transfer ability may explain the greater photocatalytic efficiency of UiO-66-CrCAT over UiO-66-GaCAT. The charge accepting ability of these trivalent ions are expected to be quite different due to differences in their outer shell electronic configurations (Cr(III) [Ar]3d3versus Ga(III) [Ar]3d10).35,36 More energy is needed to accept electrons for Ga-species because the redox potential of Ga(III)/Ga(II) is higher than Cr(III)/Cr(II). This is consistent with the ∼2× higher turnover number demonstrated by UiO-66-CrCAT when compared to UiO-66-GaCAT. Moreover, lifetimes of solid-state fluorescence, obtained by time-correlated single photon counting (TCSPC), confirmed that charge transfer between catbdc ligand and metals occurred through a LMCT mechanism (Fig. 4b, see details in ESI†). This result also suggests that UiO-66-CrCAT holds charges longer than UiO-66-GaCAT for possible electron transfer to CO2.
 | ||
| Fig. 4 (a) Photoluminescence (PL) spectra of UiO-66-CAT series; (b) solid-state fluorescent lifetime of excited states by time-correlated single photon counting (TCSPC) method. | ||
The catalytic ability with respect to turnover frequency (TOF, h−1) of the UiO-66-M(III)CAT MOFs were compared to other catalytic systems (Tables S4–S6, ESI†). The turnover frequency of UiO-66-CrCAT (1.87 h−1) and UiO-66-GaCAT (1.02 h−1) were substantially greater than many reported heterogeneous systems that produce formate or formic acid as the photoproduct (Table S4, ESI†). In contrast, the TOF of these MOFs was lower than that of many homogenous systems reported (Table S5, ESI†); however, the MOFs have the advantage of being both recyclable and not requiring an exogenous photosensitizer, which are both shortcomings of the homogenous systems reported. Therefore, the MOF catalysts reported here balance the advantages of existing heterogenous and homogenous photoreduction catalysts. In addition, the UiO-66-M(III)CAT MOFs showed good photocatalytic ability when compared to other MOF-based CO2 reduction photocatalytic systems studied to date (Table S6, ESI†). When compared to other MOFs that do not use an added exogenous photosensitizer, TOF values for the UiO-66-M(III)CAT MOFs are noticeably better than previously studied MOFs that generate formate from CO2.
New MOF CO2 reduction photocatalysts were prepared from isolated monocatecholato metal sites that were active under visible light irradiation. The catbdc substituted UiO-66-CAT generated electron–hole pairs under visible light without light sensitizers. Both UiO-66-M(III)CAT-derivatives reduced CO2 to HCOOH with the aid of BNAH and TEOA. The Cr-derivative showed better efficiency than Ga due to its open shell electronic structure. Further optimization of these systems may produce materials with the advantages of heterogeneous systems, but with activities comparable to homogenous reduction catalysts.
These experiments were supported by a grant from the Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering under Award No. DE-FG02-08ER46519 (Y. L., H. F., S. M. C.). Additional support for XPS, PL, and TCSPC studies were provided to S. K. and J. K. K. by the Korea Center for Artificial Photosynthesis (2009-0093881).
Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 CAS.
- B. Kumar, et al. , Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- P. D. Tran, L. H. Wong, J. Barber and J. S. C. Loo, Energy Environ. Sci., 2012, 5, 5902–5918 CAS.
- Y. Izumi, Coord. Chem. Rev., 2013, 257, 171–186 CrossRef CAS PubMed.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J. M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- W.-H. Wang, et al. , Energy Environ. Sci., 2012, 5, 7923–7926 CAS.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- Y. Fu, et al. , Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- Y. Horiuchi, et al. , J. Phys. Chem. C, 2012, 116, 20848–20853 CAS.
- T. Toyao, et al. , Catal. Sci. Technol., 2013, 3, 2092–2099 CAS.
- Y. Lee, S. Kim, J. K. Kang and S. M. Cohen, Chem. Commun., 2015, 51, 5735–5738 RSC.
- L. Li, et al. , Chem. Sci., 2014, 5, 3808–3813 RSC.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- H. Fei, et al. , J. Am. Chem. Soc., 2014, 136, 4965–4973 CrossRef CAS PubMed.
- H. Fei, S. Pullen, A. Wagner, S. Ott and S. M. Cohen, Chem. Commun., 2015, 51, 66–69 RSC.
- C. H. Hendon, et al. , J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed.
- H. Q. Pham, et al. , J. Phys. Chem. C, 2014, 118, 4567–4577 CAS.
- L. Shen, et al. , Phys. Chem. Chem. Phys., 2014, 17, 117–121 RSC.
- D. E. Wheeler and J. K. McCusker, Inorg. Chem., 1998, 37, 2296–2307 CrossRef CAS PubMed.
- A. J. Simaan, et al. , Chem. – Eur. J., 2005, 11, 1779–1793 CrossRef CAS PubMed.
- S. C. Ghosh, M. C. Biesinger, R. R. LaPierre and P. Kruse, J. Appl. Phys., 2007, 101, 114322 CrossRef PubMed.
- M. C. Biesinger, et al. , Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS PubMed.
- C. Pac, et al. , J. Am. Chem. Soc., 1981, 103, 6495–6497 CrossRef CAS.
- X. Q. Zhu, et al. , Chem. – Eur. J., 2003, 9, 3937–3945 CrossRef CAS PubMed.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- W. Liang, R. Babarao and D. M. D'Alessandro, Inorg. Chem., 2013, 52, 12878–12880 CrossRef CAS PubMed.
- T. Reda, C. M. Plugge, N. J. Abram and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10654–10658 CrossRef CAS PubMed.
- H. Takeda, H. Koizumi, K. Okamoto and O. Ishitani, Chem. Commun., 2014, 50, 1491–1493 RSC.
- G. Zhang, G. Kim and W. Choi, Energy Environ. Sci., 2014, 7, 954–967 CAS.
- A. Hameed, M. A. Gondal and Z. H. Yamani, Catal. Commun., 2004, 5, 715–719 CrossRef CAS PubMed.
- F. C. de Oliveira, et al. , Chem. Phys. Lett., 2013, 585, 84–88 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, supplementary figures and tables, details on comparison study. See DOI: 10.1039/c5cc04506a |
| This journal is © The Royal Society of Chemistry 2015 |