
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Significant stabilization of palladium by gold in the bimetallic nanocatalyst leading to an enhanced activity in the hydrodechlorination of aryl chlorides†
Sangita
Karanjit
a,
Atchaleeya
Jinasan
b,
Ekasith
Samsook
b,
Raghu N.
Dhital
a,
Kenichi
Motomiya
c,
Yoshinori
Sato
cd,
Kazuyuki
Tohji
c and
Hidehiro
Sakurai
*ae
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: hsakurai@chem.eng.osaka-u.ac.jp
bNANOCAST Laboratory, Center for Catalysis Department of Chemistry and Center for Innovation in Chemistry, Faculty of Science, Mahidol University, Ratchathewi, Bangkok 10400, Thailand
cGraduate School of Environmental Studies, Tohoku University, Aoba 6-6-20, Aramaki, Aoba-ku, Sendai 980-8579, Japan
dInstitute for Biomedical Sciences, Interdisciplinary Cluster for Cutting Edge Research, Shinshu University, Asahi 3-1-1, Matsumoto, 390-8621, Japan
eJapan Science and Technology Agency, ACT-C 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 22nd June 2015
Abstract
The stabilization effect of Au towards Pd changed the reactivity of Pd in Au/Pd bimetallic nanoclusters, altering the reaction mechanism from homogeneous to heterogeneous in dechlorination reaction of aryl chlorides. This phenomenon was illustrated by the observed enhancement of the rate of reaction by in situ generated Au-rich bimetallic Au/Pd nanoclusters.
Catalysis by palladium nanoparticles (NPs) has aroused great interest because of its widespread applications in organic synthesis, including C–C bond-forming reactions. The nature of the active catalytic species in reactions such as the Mizoroki–Heck1 and Suzuki–Miyaura2 coupling reactions has been an issue of great interest in the past several decades. Most of the C–C coupling reactions are found to be catalyzed by soluble palladium complexes following a usual homogenous mechanism.3,4d,5a,9 Leaching of Pd in the coupling reactions has already been well-established as demonstrated by various experimental approaches.9a It has been reported that leaching of Pd species from the surface of a catalyst through oxidative addition of aryl halides gives ArPdX species that react homogenously, forming the desired product and Pd0 species (Scheme 1). The resulting Pd0 species can react further or they can redeposit on the cluster surface.4–6 Alternatively, in the absence of a strong ligand or stabilizer, they can be deactivated through the formation of Pd black.4 It is a major limitation of Pd NP chemistry in coupling reactions because once oxidative addition occurs, Pd2+ will form and it will no longer stay on the surface. Stabilization of such active palladium species either in solution or on a heterogeneous surface is a quite difficult task. For the last few decades, researchers have been paying much attention to developing new strategies for designing high performance heterogeneous catalysts to prevent the usual homogenous coupling mechanism by developing immobilizing protocols.7 Recent publications on coupling reactions utilizing catalysts such as Pd grafted onto mesoporous materials7a,b,f or zeolites7c and Pd on HAP macroligands7d,e claim to proceed through heterogenous mechanism. However, such strategies are limited and a new and simple strategy to develop an efficient heterogeneous catalytic system that can promise to prevent leaching, agglomeration and deactivation of catalysts is essential.
 | ||
| Scheme 1 Proposed oxidative addition mechanism on Pd and Au/Pd catalysts. | ||
Bimetallic nanoparticles have attracted attention because of their enhanced catalytic activities, selectivities, and stabilities in comparison to their single-metal counterparts.10 Especially, the Au/Pd bimetallic alloy has superior catalytic activities in various types of reactions under mild conditions.11–17 The activity and selectivity of the Pd catalyst can be enhanced by alloying with Au due to the synergistic effects of bimetallic surfaces,16d,e such as ensemble and the ligand effects.10b Furthermore, bimetallization can result in stabilization of nanoparticles in cases where one metal acts as a stabilizer for the other, preventing aggregation.10b,16e,f
In our previous report, we observed unique catalytic activity of poly(N-vinylpyrrolidone) (PVP)-stabilized Au/Pd bimetallic nanoparticles17 for C–Cl bond activation in Ullmann coupling reactions of chloroarenes17a and the unusual Suzuki–Miyaura-type coupling reaction.17b Through DFT calculation of the reaction mechanism, we realized that spillover of Cl over gold is a crucial step in the reaction, which drives the reaction through the heterogeneous path preventing the homogeneous path (Scheme 1).17a,18 The best conditions for activation of C–Cl bonds for C–C bond-formation reactions involved the use of N,N-dimethylformamide (DMF) as solvent and a hydrogen donor (Scheme 2, conditions a; Table 1, entry 1). So, we expected that the use of other reducing solvents, such as alcohols (Scheme 2; conditions b), would result in selective activation of the C–Cl bond towards hydrodechlorination. Similar alteration of product selectivity of the two competing processes based on concentration of a reductant has been well discussed by Sasson and coworkers.8
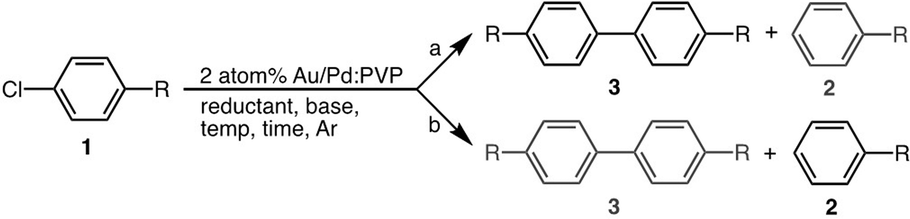 | ||
Scheme 2 C–Cl bond activation by the Au/Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PVP catalyst. :PVP catalyst. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | t (h) | Catalyst | Yieldb (%) | |
| 2 | 3 | ||||
| a Reaction conditions: 1 (0.1 mmol), catalyst (2 atom%), KOH (150 mol%), solvent (3 mL), 45 °C, in argon. b Yield by GC analysis. c At 25 °C. d 1 atom%. | |||||
| 1 | DMF–H2O (1:2) |
24 | Au/Pd:PVP |
6 | 92 |
| 2 | DMSO–H2O (1:1) |
24 | Au/Pd:PVP |
0 | 0 |
| 3 | MeOH–H2O (1:1) |
24 | Au/Pd:PVP |
22 | 20 |
| 4 | EtOH–H2O (1:1) |
24 | Au/Pd:PVP |
63 | 14 |
| 5 | PrOH–H2O (1:1) |
24 | Au/Pd:PVP |
70 | 17 |
| 6 | EtOH | 24 | Au/Pd:PVP |
78 | 11 |
| 7 | iPrOH | 1 | Au/Pd:PVP |
>95 | Trace |
| 8c | iPrOH | 5 | Au/Pd:PVP |
>95 | Trace |
| 9 | iPrOH | 1 | Au:PVP |
0 | 0 |
| 10 | iPrOH | 1 | Pd:PVP |
24 | 0 |
| 11 | iPrOH | 1 | Au:PVPd + Pd:PVPd |
51 | Trace |
In the course of our study to elucidate the stabilizing effect of gold toward palladium, we chose the hydrodechlorination reaction as our model reaction because, for Pd, it is difficult to activate C–Cl bonds and once it is activated, Pd may be released as a Pd(II) ion in solution due to the oxidative addition process (Scheme 1). For this species, the second oxidative addition process is very difficult and it is quite impossible to get the Ullmann coupling product due to low nucleophilicity on Pd and strong electrophilicity of C–Cl bonds.17a Instead, if we use the hydride source, it will possibly undergo reduction, giving arene as the product even through the usual homogenous mechanism and release the Pd0 ion which can be captured by Au as a stabilizing ligand. Au is the best choice among other metals to capture Pd because of a similar lattice constant and low energy demand for bimetallization.16e–h Besides, the nucleation process of Pd is relatively slow.16i In this work, we attempt to release Pd and trap it by Au during hydrodechlorination reaction and tune the reaction mechanism from homogenous to heterogeneous.
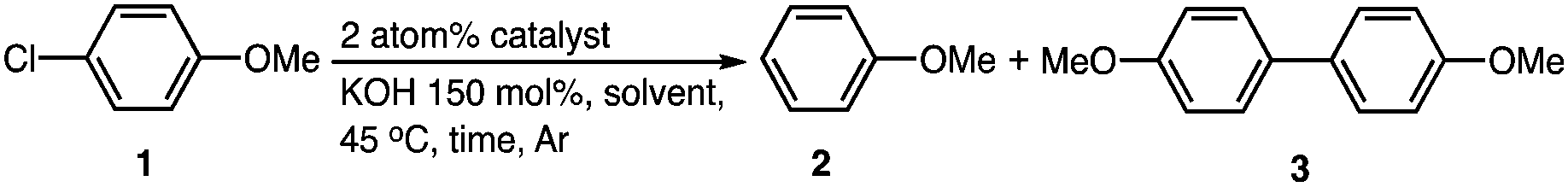
First, to identify optimal conditions for the dechlorination, the reaction of 1-chloro-4-methoxybenzene (1) was carried out in the presence of Au/Pd:PVP as a catalyst and potassium hydroxide as the base in dimethyl sulfoxide (DMSO), no reaction occurred because of the nonreducing nature of the solvent (entry 2). However, the yield of dechlorination product methoxybenzene (2) increased in alcohol solvents. Reactions in 1:1 mixtures of methanol, ethanol, or propan-2-ol with water gave 2 in yields of 22%, 63%, and 70%, respectively (entries 3–5) and without water in ethanol (78%) and propanol (>95%) (entries 6 and 7). The best conditions involved the use of anhydrous propan-2-ol as the sole solvent; under these conditions, the reaction was completed within one hour at 45 °C and five hours at 25 °C giving >95% yield of 2 (entries 7 and 8). The optimized or slightly modified reaction conditions were applied to the dechlorination of various substrates including polychlorinated compounds and the results are summarized in Table S1 (ESI†).
The efficiency of various Au and Pd catalysts for the dechlorination reaction was compared by performing the reaction of 1-chloro-4-methoxybenzene (1) for 1 h (Table 1). The highest catalytic activity was shown by Au/Pd bimetallic alloy nanoparticles, which gave 2 quantitatively (Table 1, entry 7). The dechlorination reaction of 1 did not proceed when Au:PVP was used as a catalyst (entry 9), whereas Pd:PVP gave 2. When Pd:PVP was used as a catalyst under the same conditions, the yield of 2 was only 24% (entry 10), while the physical mixture of the two monometallic catalysts Au:PVP and Pd:PVP (Au + Pd) gave 51% yield, which was higher than that obtained with Pd:PVP alone (entry 11). To test the effect of Au on the catalytic activity of Pd, we performed the reaction under identical conditions using physical mixtures of the two monometallic catalysts in varying Au and Pd ratios (Fig. S1, ESI†). Even a small amount of Pd in Au, or vice versa, enhanced the catalytic activity to a value higher than that displayed by Au or Pd alone.
Measurements of the kinetics of consumption of 1 (Fig. 1) showed differences in the catalytic activities of Au/Pd alloy, Pd, and (Au + Pd) nanoparticles. The reaction with bimetallic Au/Pd:PVP was a very fast second-order reaction with a rate constant for consumption of 1 of k = 5.6 × 10−1 L mol−1 h−1 (Fig. S2, ESI†). The reaction with monometallic Pd:PVP, on the other hand, was very slow and showed first-order kinetics. The 1:1 physical mixture of Au:PVP and Pd:PVP show a reactivity that was intermediate between those of the two monometallic catalysts. At the beginning of the reaction, the catalytic activity of Pd:PVP (2 atom%) was higher, and the reaction rate was almost double than that observed with the Au:PVP (1 atom%) + Pd:PVP (1 atom%) mixture. However, after two hours, the activity of the Au + Pd mixture gradually increased in comparison to that of Pd alone. The kinetics of consumption of 1 in the presence of the Au + Pd mixture at 25 °C showed a linear fit to the first-order plot of −ln[A] versus time, with an observed rate of consumption of 1 (k1a) = 3.7 × 10−2 h−1 for the initial 2 hours of reaction. However, the slope then increased significantly, corresponding to an increase in the rate to 4.6 × 10−2 h−1 and a change in the order of the reaction (Fig. 1b).
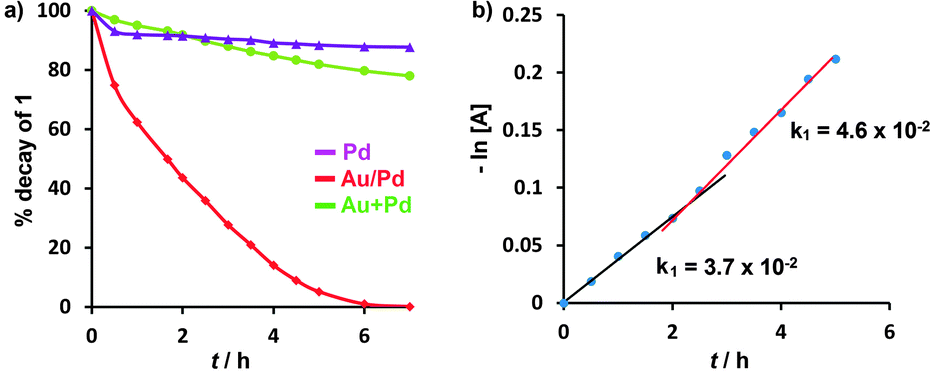 | ||
| Fig. 1 (a) Time-dependent consumption of 1 at 25 °C; reaction conditions: 1 (0.3 mmol), NaOH (150 mol%), catalyst (2 atom%), iPrOH; (b) plot of −ln[A] vs. time for Au + Pd. | ||
On the basis of the above results, we hypothesized that leaching might occur from the surface during catalysis by monometallic Pd due to oxidative addition of the C–Cl bond. This species must be exclusively responsible for the initial catalytic activity through a homogeneous mechanism because Au:PVP on its own shows no activity in the reaction. In the presence of gold, in situ reduction of Pd(II) under the reaction conditions might lead to the Au/Pd bimetallic catalyst with high dispersion of Pd on Au (Table 1, entry 11). We therefore propose the existence of two catalytic cycles for the observed reactivities of monometallic and bimetallic nanoparticles, respectively (Scheme 3). The catalytic reaction involving the bimetallic nanoparticles follows a heterogeneous mechanism and occurs on the surface of the catalyst (cycle A) because of the spillover of Cl over gold. The process is very fast, with no aggregation or deactivation of the catalyst at any stage of the reaction. In the case of monometallic Pd:PVP in the absence of Au, the catalyst shows a homogeneous reaction mechanism (cycle B) through leached Pd. This is a slow process and, as the reaction proceeds, the Pd0 species becomes gradually deactivated through aggregation, with the formation of Pd black. In the presence of Au, however, the leached palladium species that are present in solution are captured by Au to form bimetallic Au/Pd:PVP and both catalytic cycles operating simultaneously in the case of physical mixtures of Au:PVP and Pd:PVP.
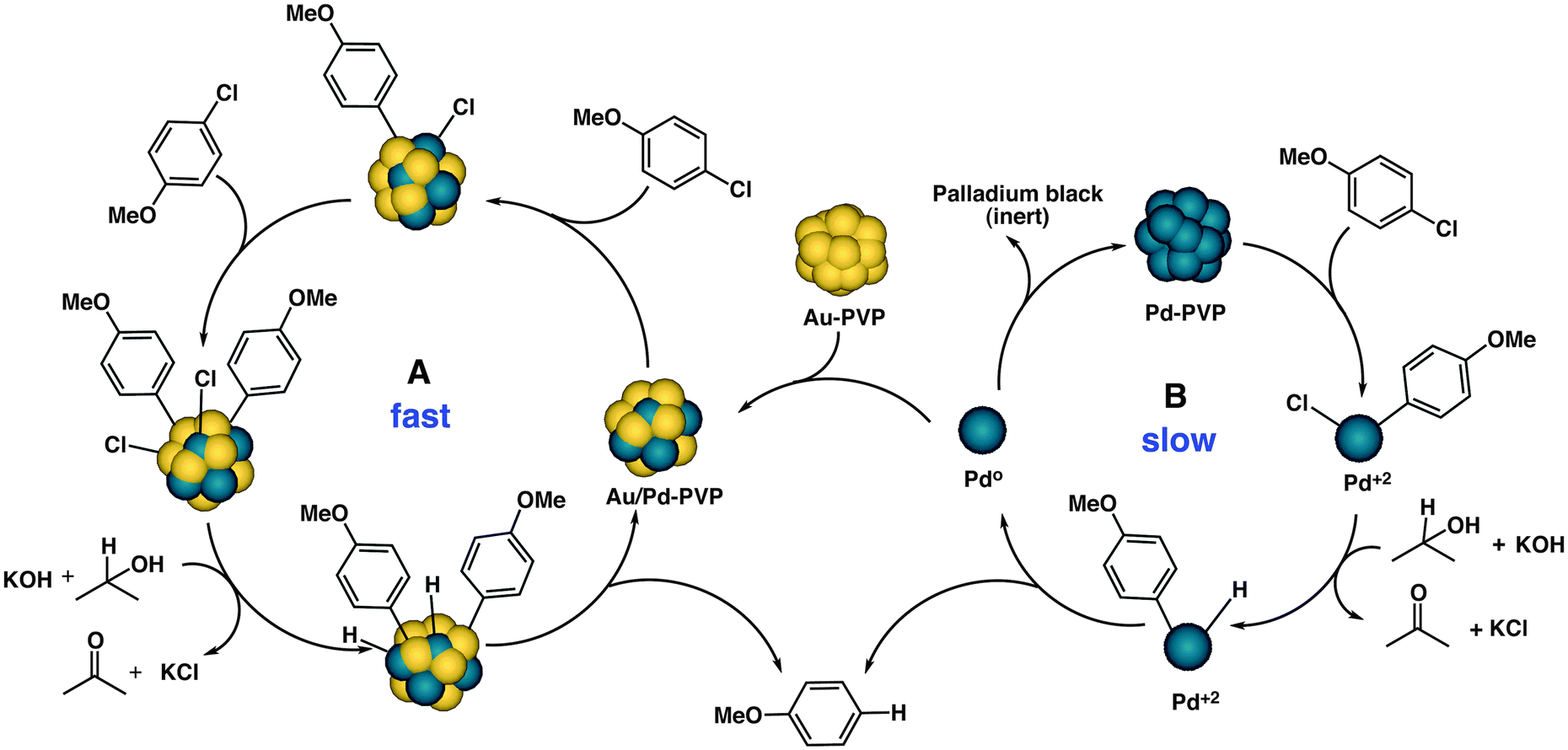 | ||
| Scheme 3 Proposed catalytic cycles A (heterogeneous) and B (homogeneous) for the dechlorination reaction. | ||
To verify our hypothesis, we used transmission electron microscopy (TEM) to examine structural and morphological changes in the metal nanoparticle as a result of the reaction. The two monometallic nanoparticles Au:PVP and Pd:PVP were mixed in a 1:1 ratio, and TEM images were recorded before and after the reaction (Fig. S3 and S4, ESI†). The images taken before reaction showed the Au:PVP and Pd:PVP nanoparticles separately, with average sizes of 1.6 ± 0.4 nm and 3.9 ± 0.6 nm, respectively. However, images recorded after the reaction showed morphological changes, with a growth in the size of the nanoparticles. Detailed scanning transmission electron microscopy (STEM) energy dispersive X-ray spectroscopy (EDX) measurements showed that most of the particles are polymorphological with non-uniform lattice patterns. However, the existence of some of the bimetallic nanoparticles containing various amounts of Au and Pd (Table S2, Fig. S5, ESI†) is revealed. These particles show a uniform lattice pattern and single crystallinity but possess two metals (Fig. 2). This supports our belief that the enhanced catalytic activity of Pd is due to the dilution of surface Pd atoms by gold in forming these very few numbers of Au rich Au/Pd bimetallic nanoparticles in situ.
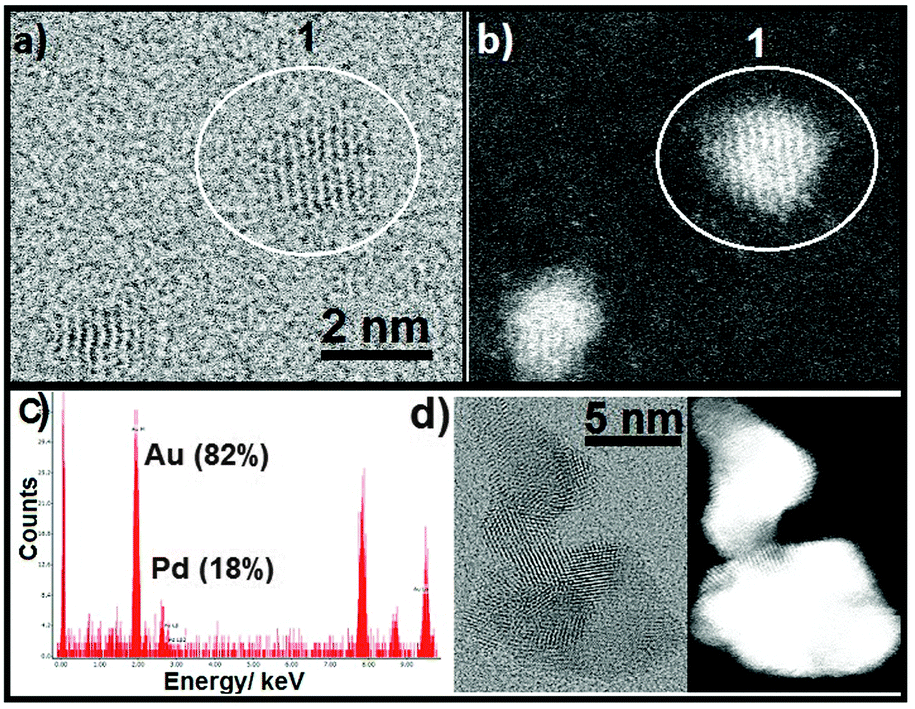 | ||
| Fig. 2 (a) Bright-field and (b) high-angle annular dark-field (HAADF) STEM images of particle 1 after reaction. (c) STEM-EDX spectrum of particle 1. (d) Bright-field (left) and HAADF STEM images of a polymorphic particle formed after reaction. | ||
In summary, through hydrodechlorination reaction results we could clearly conclude the leaching of Pd species; these enhance the catalytic activity by interacting with the Au nanoparticles in Au/Pd bimetallic nanoparticles generated in situ and the heteroatomic gold drives the reaction mechanism from homogeneous to heterogeneous. This phenomenon provides a good illustration of a bimetallic effect in which one metal dopes another metal. We also found that Au plays two important roles. The first is to capture and stabilize Pd, and the second is to produce a bimetallic system that catalyzes the reaction by a different mechanism.
Notes and references
- (a) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320 CrossRef CAS; (b) K. Mori, T. Mizoroki and A. Ozaki, Bull. Chem. Soc. Jpn., 1973, 46, 1505 CrossRef CAS; (c) R. F. Heck, Acc. Chem. Res., 1979, 12, 146 CrossRef CAS.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) F. Fernandez, B. Cordero, J. Durand, G. Muller, F. Malbosc, Y. Kihn, E. Teumaa and M. Gomez, Dalton Trans., 2007, 5572 RSC; (c) D. Sanhes, E. Raluy, S. Retory, N. Saffon, E. Teumaa and M. Gomez, Dalton Trans., 2010, 9719 RSC.
- (a) N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef CAS; (b) R. L. Augustine and S. T. O’Leary, J. Mol. Catal. A: Chem., 1995, 95, 277 CrossRef CAS; (c) R. L. Augustine and S. T. O’Leary, J. Mol. Catal., 1992, 72, 229 CrossRef CAS; (d) S. Jansat, J. Durand, I. Favier, F. Malbosc, C. Pradel, E. Teuma and M. Gomez, ChemCatChem, 2009, 1, 244 CrossRef CAS; (e) L. Rodriguez-Perez, C. Pradel, P. Serp, M. Gomez and E. Teuma, ChemCatChem, 2011, 3, 749 CrossRef CAS.
- (a) M. T. Reetz and E. Westermann, Angew. Chem., Int. Ed., 2000, 39, 165 CrossRef CAS; (b) M. T. Reetz, G. Lohmer and R. Schwickardi, Angew. Chem., Int. Ed., 1998, 37, 481 CrossRef CAS; (c) A. H. M. de Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Hendrickx and J. G. de Vries, Org. Lett., 2003, 5, 3285 CrossRef CAS PubMed; (d) M. B. Thathagar, J. E. ten Elshof and G. Rothenberg, Angew. Chem., Int. Ed., 2006, 45, 2886 CrossRef CAS PubMed; (e) L. D. Pachon and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288 CrossRef; (f) J. Durand, E. Teuma and M. Gómez, Eur. J. Inorg. Chem., 2008, 3577 CrossRef CAS; (g) I. Favier, D. Madec, E. Teuma and M. Gomez, Curr. Org. Chem., 2011, 15, 3127 CrossRef CAS.
- (a) A. V. Gaikwad, A. Holyuigre, M. B. Thathagar, J. E. ten Elshof and G. Rothenberg, Chem. – Eur. J., 2007, 13, 6908 CrossRef CAS PubMed; (b) S. Mac-Quarrie, J. H. Horton, J. Barnes, K. McEleney, H. P. Loock and C. M. Crudden, Angew. Chem., Int. Ed., 2008, 47, 3279 CrossRef CAS PubMed; (c) K. Kohler, R. G. Heidenreich, S. S. Soomro and S. S. Prockl, Adv. Synth. Catal., 2008, 350, 2930 CrossRef; (d) K. Q. Yu, W. Sommer, J. M. Richardson, M. Weck and C. W. Jones, Adv. Synth. Catal., 2005, 347, 161 CrossRef CAS.
- (a) S. Higashibayashi and H. Sakurai, Chem. Lett., 2007, 36, 18 CrossRef CAS; (b) A. F. G. Masud Reza, S. Higashibayashi and H. Sakurai, Chem. – Asian J., 2009, 4, 1329 CrossRef PubMed; (c) S. Higashibayashi, A. F. G. Masud Reza and H. Sakurai, J. Org. Chem., 2010, 75, 4626 CrossRef CAS PubMed; (d) M. Yamanaka, M. Morishima, Y. Shibata, H. Higashibayashi and H. Sakurai, Organometallics, 2014, 33, 3060 CrossRef CAS.
- (a) N. Hoshiya, M. Shimoda, H. Yoshikawa, S. Shuto and M. Arisawa, J. Am. Chem. Soc., 2010, 132, 7270 CrossRef CAS PubMed; (b) B. Yuan, Y. Pan, Y. Li, B. Yin and H. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4054 CrossRef CAS PubMed; (c) L. Djakovitch and K. Koehler, J. Chem. Soc., 2001, 123, 5990 CrossRef CAS; (d) K. Mori, K. Yamaguchi, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Chem. Soc., 2002, 124, 11572 CrossRef CAS; (e) K. Mori, T. Hara, M. Oshiba, T. Mizugaki, K. Ebitani and K. Kaneda, New J. Chem., 2005, 29, 1174 RSC; (f) R. J. White, R. Luque, V. L. Budarin and J. H. Clark, Chem. Soc. Rev., 2009, 38, 481 RSC; (g) S. Sá, M. B. Gawande, A. Velhinho, J. P. Veiga, N. Bundaleski, J. Trigueiro, A. Tolstogouzov, O. M. N. D. Teodoro, R. Zboril, R. S. Varma and P. S. Branco, Green Chem., 2014, 16, 3494 RSC.
- S. Mukhopadhyay, G. Rothenberg, D. Gitis, H. Wiener and Y. Sasson, J. Chem. Soc., Perkin Trans. 2, 1999, 2481 RSC.
- (a) J. M. Richardson and C. W. Jones, Adv. Synth. Catal., 2006, 348, 1207 CrossRef CAS; (b) M. B. Thathagar, P. J. Kooyman, R. Boerleider, E. Jansen, C. J. Elsevier and G. Rothenberg, Adv. Synth. Catal., 2005, 347, 1965 CrossRef CAS; (c) S. S. Prockl, W. Kleist, M. A. Gruber and K. Kohler, Angew. Chem., Int. Ed., 2004, 43, 1881 CrossRef PubMed.
- (a) C. W. Yi, K. Luo, T. Wei and D. W. Goodman, J. Phys. Chem. B, 2005, 109, 18535 CrossRef CAS PubMed; (b) F. Gao and D. W. Goodman, Chem. Soc. Rev., 2012, 41, 8009 RSC.
- (a) J. Xu, T. White, P. Li, C. H. He, J. G. Yu, W. K. Yuan and Y.-F. Han, J. Am. Chem. Soc., 2010, 132, 10398 CrossRef CAS PubMed; (b) T. Gracia, S. Agouram, A. Dejoz, J. F. Sanchez-Royo, L. Torrente-Murciano and B. Solsona, Catal. Today, 2015, 248, 48 CrossRef; (c) M. O. Nutt, K. N. Heck, P. Alvarez and M. S. Wong, Appl. Catal., B, 2006, 69, 115 CrossRef CAS; (d) A. Sárkány, O. Geszti and G. Sáfrán, Appl. Catal., A, 2008, 350, 157 CrossRef; (e) N. E. Kolli, L. Delannoy and C. J. Louis, Catalysis, 2013, 297, 79 CrossRef CAS; (f) T. Longfei, W. Xiaoli, C. Dong, L. Huiyu, M. Xianwei and T. Fangqiong, J. Mater. Chem. A, 2013, 1, 10382 RSC; (g) J. Long, H. Liu, S. Wu, S. Liao and Y. Li, ACS Catal., 2013, 3, 647 CrossRef CAS.
- (a) X. Wei, X. F. Yang, A. Q. Wang, L. Li, X. Y. Liu and T. Zhang, J. Phys. Chem., 2012, 116, 6222 CAS; (b) S. Carrettin, P. McMorn, P. Johnston, K. Griffin, C. J. Kielyc and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1329 RSC.
- N. Toshima, J. Macromol. Sci., Chem., 1990, 27, 1225 CrossRef.
- M. Chen, D. Kumar, C.-W. Yi and D. W. Goodman, Science, 2005, 310, 291 CrossRef CAS PubMed.
- H. Zhang, T. Watanabe, M. Okumura, M. Haruta and N. Toshima, Nat. Mater., 2012, 11, 49 CrossRef PubMed.
- (a) N. K. Chaki, H. Tsunoyama, Y. Nigishi, H. Sakurai and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 4885 CrossRef CAS; (b) S. S. Yudha, R. N. Dhital and H. Sakurai, Tetrahedron Lett., 2011, 52, 2633 CrossRef; (c) O. Sophiphun, J. Wittayakun, R. N. Dhital, S. Haesuwannakij, A. Murugadoss and H. Sakurai, Aust. J. Chem., 2012, 65, 1238 CrossRef CAS; (d) A. Murugadoss, K. Okumura and H. Sakurai, J. Phys. Chem. C, 2012, 116, 26776 CrossRef CAS; (e) D. Wang, A. Villa, F. Porta, L. Prati and D. Su, J. Phys. Chem. C, 2008, 112, 8617 CrossRef CAS; (f) C. Rossy, J. Majimel, E. Fouquet, C. Delacôte, M. Boujtita, C. Labrugère, M. Tréguer-Delapierre and F. X. Felpin, Chem. – Eur. J., 2013, 19, 14024 CrossRef CAS PubMed; (g) H. B. Liu, U. Pal, A. Medina, C. Maldonado and J. A. Ascencio, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 075403 CrossRef; (h) J. Cai and Y. Y. Ye, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 8398 CrossRef CAS; (i) M. L. Wu, D. H. Chen and T. C. Huang, Langmuir, 2001, 17, 3877 CrossRef CAS.
- (a) R. N. Dhital, C. Kamonsatikul, E. Somsook, K. Bobuatong, M. Ehara, S. Karanjit and H. Sakurai, J. Am. Chem. Soc., 2012, 134, 20250 CrossRef CAS PubMed; (b) R. N. Dhital and H. Sakurai, Chem. Lett., 2012, 41, 630 CrossRef CAS.
- B. Boekfa, E. Pahl, N. Gaston, H. Sakurai, J. Limtrakul and M. Ehara, J. Phys. Chem. C, 2014, 118, 22188 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc04432d |
| This journal is © The Royal Society of Chemistry 2015 |