
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzymatic synthesis and post-functionalization of two-dimensional crystalline cellulose oligomers with surface-reactive groups†
Yusuke
Yataka
,
Toshiki
Sawada
and
Takeshi
Serizawa
*
Department of Organic and Polymeric Materials, Tokyo Institute of Technology, 2-12-1-H121 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: serizawa@polymer.titech.ac.jp
First published on 26th June 2015
Abstract
Two-dimensional (2D) nanomaterials have gained considerable attention due to their unique structural and physicochemical properties. Herein, we synthesized 2D crystalline cellulose oligomers with surface-reactive azide groups through enzymatic reactions and covalently post-functionalized them with 1-ethynyl pyrene through copper(I)-catalyzed Huisgen cycloaddition reactions.
Motivated by the development of 2D inorganic nanomaterials, such as graphene,1 layered metal chalcogenides,2,3 and layered metal oxide,3 2D “organic” nanomaterials have gained considerable attention due to their unique properties, such as lightweight, structural control and flexibility, and tailored reactivities.4 2D organic nanomaterials are synthesized either by covalent or non-covalent approaches using well-defined building blocks and include 2D polymers,5 covalent organic frameworks,6 and supramolecular and/or crystalline organic layers.7 They typically have sizes with thicknesses of sub-ten nanometers, which are several orders of magnitude smaller than the lateral dimensions. Due to their porous structure and physicochemical properties they have potential application in the fields of membranes, storage, sensing, catalysis, and devices.4 Precise synthesis and subsequent functionalization of regularly structured 2D organic nanomaterials are meaningful and challenging targets.
Nanocellulose has recently been focused on as a naturally abundant sustainable nanomaterial due to its unique morphological, mechanical, and chemical properties.8 To avoid too much dependence on natural resources, organic9 and enzymatic10,11 syntheses of cellulose have been investigated. Such processes are expected to allow for better control of the chemical and crystalline structures. The enzymatic process is significant for the one-step sustainable synthesis of cellulose oligomers (so-called “cellodextrin”) under aqueous and mild conditions and is typically performed by glycosylation reactions using cellulase11 and cellodextrin phosphorylase (CDP),12–15 respectively. 2D crystalline cellulose oligomers with sheet-like morphologies were successfully synthesized by the latter phosphorolytic reactions. In particular, when α-D-glucose 1-phosphate (αG1P) monomers were propagated to β-D-glucose primers by CDP isolated from Clostridium thermocellum, 2D crystalline cellulose oligomers with 4.5 nm thickness were obtained.15 The synthesized oligomers had an average degree of polymerization (DP) of 9 and formed the anti-parallel cellulose II allomorph, in which the oligomers were aligned perpendicular to the nanomaterial surface, thus endowing lamella structures. Therefore, the termini of the cellulose oligomers could be periodically accumulated on the nanomaterial surface.
Considering previous knowledge on the phosphorolytic synthesis of cellulose oligomers,12,14,15 CDP has poor recognition capability against anomeric substitutes of primers. In fact, 4-O-β-D-glycosyl-D-altrose, sophorose, and laminaribiose were also applied to the phosphorolytic reactions for the propagation of cellulose chains,12,14 similarly to cellobiose14 and β-D-glucose.15 Therefore, we hypothesized that diverse design and synthesis of surface-functionalized 2D crystalline cellulose oligomers would be achieved using β-D-glucose derivative primers with the desired anomeric substitutes. Herein, 2D crystalline cellulose oligomers with surface-reactive azide groups were synthesized by applying 1-azide-1-deoxy-β-D-glucopyranoside (β-glucosyl azide) primers to the CDP-catalyzed phosphorolytic reaction with αG1P monomers (Fig. 1a), and subsequently, as a proof-of-concept for post-functionalization, 1-ethynyl pyrene was covalently conjugated to the azide groups through copper(I)-catalyzed Huisgen cycloaddition reactions (Fig. 1b). Environmentally friendly and one-step synthetic processes under aqueous and mild conditions were used to synthesize 2D surface-reactive nanomaterials for post-functionalization with small organic molecules.
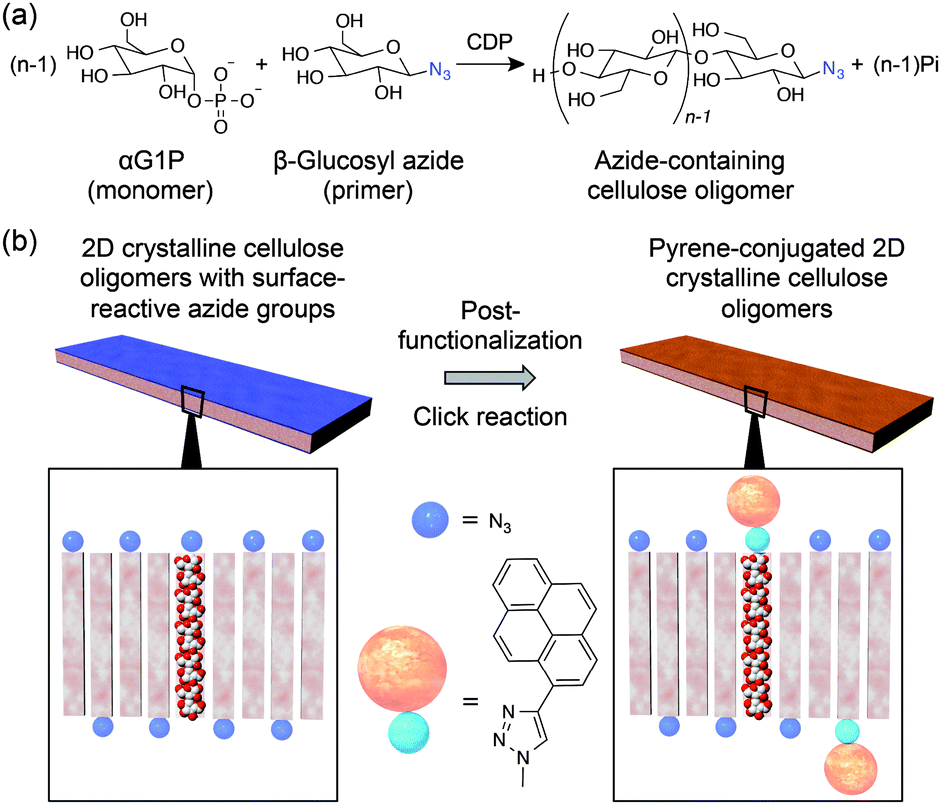 | ||
| Fig. 1 (a) Synthetic scheme of cellulose oligomers with azide groups at the reductive end through enzymatic reactions by CDP using αG1P monomers and β-glucosyl azide primers. (b) Schematic illustration of the post-functionalization of surface-azidized 2D crystalline cellulose oligomers with 1-ethynyl pyrene through copper(I)-catalyzed Huisgen cycloaddition reactions. | ||
When αG1P (200 mM) and β-glucosyl azide (50 mM) were incubated with CDP (0.2 U mL−1) in 500 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer solutions (pH 7.5) at 60 °C for 3 days, colorless dispersions composed of water-insoluble products were obtained (Fig. S1, ESI†) (Experimental details are summarized in the ESI†). 1H nuclear magnetic resonance (NMR) spectra of the product dissolved in 4% NaOD–D2O showed signals assigned to repeating glucose units (Fig. S2, ESI†). Compared with the 1H NMR spectrum for cellulose oligomers synthesized using reference D-glucose primers instead of β-glucosyl azide under the same conditions (Fig. S3, ESI†), peaks corresponding to anomeric carbons were not observed for the product. Attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectra of the product showed a peak at 2120 cm−1 assigned to the stretching vibration band of the azide groups, whereas the other typical peaks were almost the same as those for cellulose oligomers (Fig. 2a and Fig. S4, ESI†). These observations suggested successful synthesis of oligomeric cellulose derivatives with azide groups at the reducing end. The average DP of the product was approximately 10 based on the elementary analysis (Table S1, ESI†). The conversion of αG1P was estimated to be 30% using the collected amount and average molecular weight of the product.
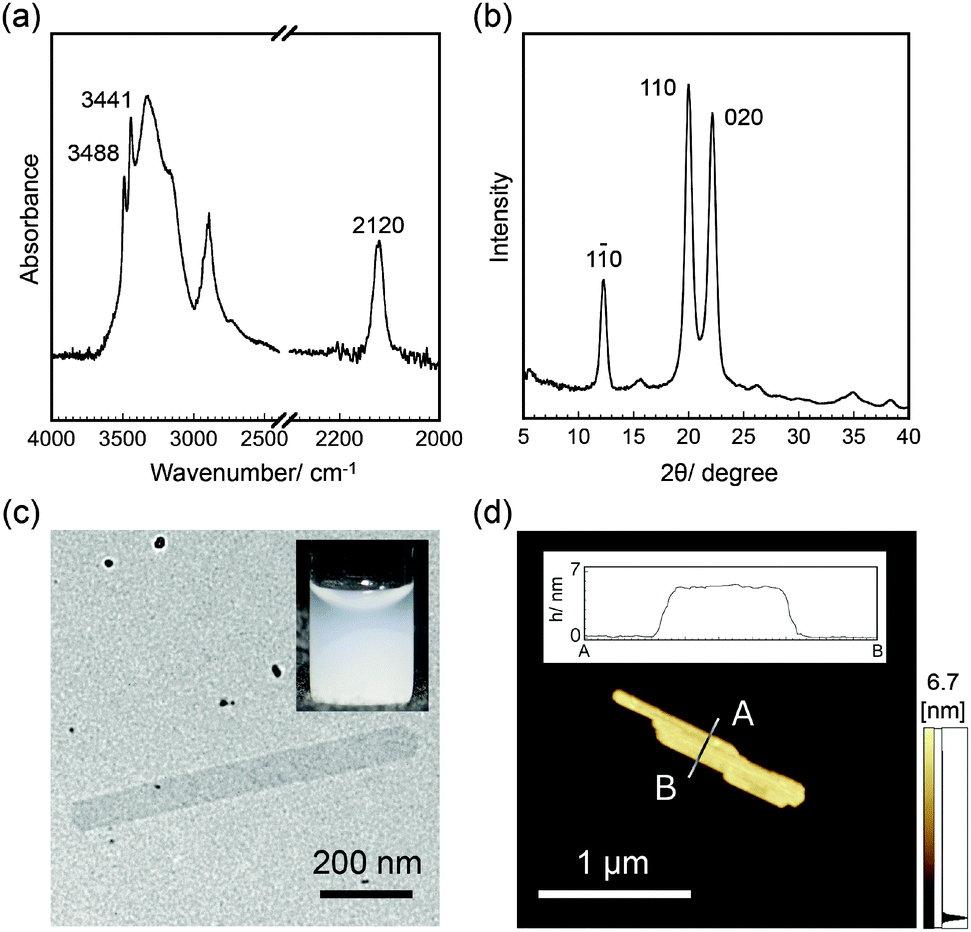 | ||
| Fig. 2 (a) ATR-FTIR spectra, (b) WAXD profiles, (c) TEM images, and (d) AFM images of 2D crystalline cellulose oligomers with surface-reactive azide groups. The insets of (c) and (d) show the dispersion in DMF and the cross-sectional analysis, respectively. | ||
Wide-angle X-ray diffraction (WAXD) measurements of the product showed diffractions assigned to d-spacings of 0.721, 0.444, and 0.401 nm, which corresponded to (1 ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0), (1 1 0), and (0 2 0) of the cellulose II allomorph, respectively (Fig. 2b and Fig. S5, ESI†).15 In addition, the sharp peaks of OH vibration bands at 3441 and 3488 cm−1 in the ATR-FTIR spectra supported the formation of the cellulose II allomorph (Fig. 2a).16 Transmission electron microscopy (TEM) observations revealed sheet-like morphologies with several hundreds of nanometer to several micrometer lengths and several hundreds of nanometer widths (Fig. 2c). Atomic force microscopy (AFM) observations confirmed the 2D nanostructures with an average thickness of 5.5 ± 0.5 nm (Fig. 2d). The thickness was consistent with the chain length of cellodecaose in the cellulose II allomorph (5.2 nm, see Fig. S6, ESI†), strongly suggesting that the cellulose chains were aligned perpendicular to the nanomaterial surface. These observations indicated that 2D crystalline cellulose oligomers with surface-reactive azide groups were successfully synthesized by CDP-catalyzed phosphorolytic reactions using αG1P monomers and β-glucosyl azide primers.
0), (1 1 0), and (0 2 0) of the cellulose II allomorph, respectively (Fig. 2b and Fig. S5, ESI†).15 In addition, the sharp peaks of OH vibration bands at 3441 and 3488 cm−1 in the ATR-FTIR spectra supported the formation of the cellulose II allomorph (Fig. 2a).16 Transmission electron microscopy (TEM) observations revealed sheet-like morphologies with several hundreds of nanometer to several micrometer lengths and several hundreds of nanometer widths (Fig. 2c). Atomic force microscopy (AFM) observations confirmed the 2D nanostructures with an average thickness of 5.5 ± 0.5 nm (Fig. 2d). The thickness was consistent with the chain length of cellodecaose in the cellulose II allomorph (5.2 nm, see Fig. S6, ESI†), strongly suggesting that the cellulose chains were aligned perpendicular to the nanomaterial surface. These observations indicated that 2D crystalline cellulose oligomers with surface-reactive azide groups were successfully synthesized by CDP-catalyzed phosphorolytic reactions using αG1P monomers and β-glucosyl azide primers.
Functionalization of 2D nanomaterials with fluorescent dye molecules opens new opportunities for imaging, sensing, and device applications.4 To perform the post-functionalization of the present 2D reactive nanomaterials based on versatile chemical reactions, 1-ethynyl pyrene, as a model of fluorescent dye molecules, was conjugated with the azide groups through copper(I)-catalyzed Huisgen cycloaddition reactions (Fig. 1b). Although the 2D crystalline cellulose oligomers were enzymatically synthesized in aqueous solutions, they were stably dispersed, even in various organic solvents. In fact, solvents were readily exchanged by centrifugation/redispersion processes. This solvent-dispersible capability is a great advantage to effectively post-functionalize them with water-insoluble hydrophobic molecules through chemical reactions in desired solvents. Therefore, DMF was selected for the click reaction because it is a good solvent for 1-ethynyl pyrene (also see the inset of Fig. 2c for the dispersion of the 2D crystalline cellulose oligomers in DMF).
When the 2D crystalline cellulose oligomers (0.45% (w/v)) were incubated with 1-ethynyl pyrene (4.5 mM) in DMF in the presence of copper(II) sulfide (0.45 mM) and ascorbic acid (1.1 mM) at ambient temperature for 1 day under a nitrogen atmosphere, an orangish product that was well-dispersed in DMF was obtained (the inset of Fig. 3a). The orangish color could not be removed even after washing the 2D crystalline cellulose oligomers with DMF by centrifugation-based solvent exchange. The reaction in the absence of copper(II) sulfide did not result in a color change. The ATR-FTIR spectra of the product showed peaks at 710 and 835 cm−1 assigned to the C–H deformation vibration bands for pyrene units (Fig. S7, ESI†).17 The TEM images (Fig. S8, ESI†) and OH vibration bands of the ATR-FTIR spectra (Fig. S7, ESI†) for the product indicated the preservation of the 2D structures and the cellulose II allomorph, even after the click reaction, respectively, suggesting that the 2D crystalline cellulose oligomers were stable platforms for click reactions. Elementary analysis estimated the percent amount of 1-ethynyl pyrene conjugated to the total cellulose oligomers to be approximately 30% under the present synthetic conditions (Table S2, ESI†). Accordingly, it was found that pyrene was successfully conjugated to the 2D crystalline cellulose oligomers through copper(I)-catalyzed Huisgen cycloaddition reactions.
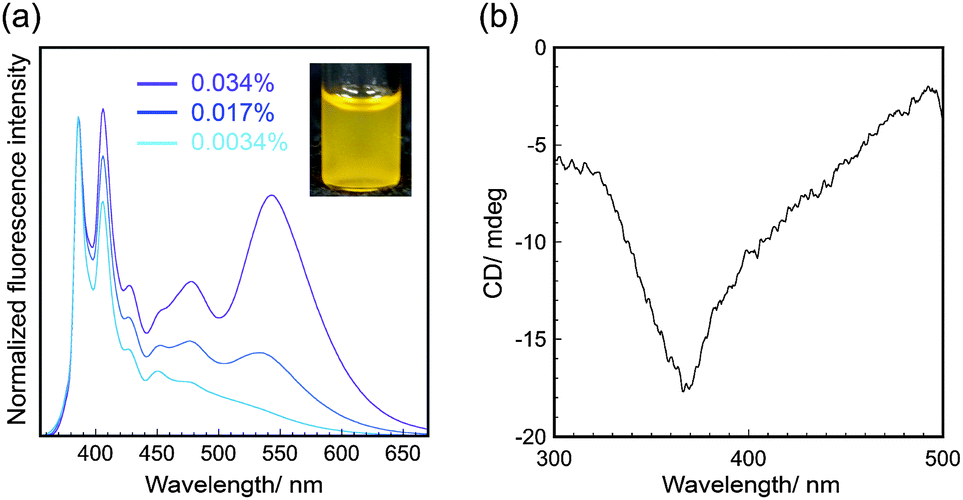 | ||
| Fig. 3 (a) Normalized fluorescence spectra (λex = 343 nm) and (b) CD spectra of pyrene-conjugated 2D crystalline cellulose oligomers dispersed in DMF. The fluorescence intensity shown in (a) was normalized based on the intensity at 386 nm. | ||
The pyrene-conjugated 2D crystalline cellulose oligomers were then characterized spectroscopically. The ultraviolet-visible (UV-Vis) absorption spectra in DMF (0.0034% (w/v)) showed typical peaks for pyrene units at 330–400 nm (Fig. S9, ESI†),18 confirming the presence of pyrene molecules on the nanomaterial surface. The fluorescence spectra obtained upon excitation at 343 nm in DMF (0.0034% (w/v)) showed two main peaks at 386 and 406 nm assigned to the emission of pyrene monomers (Fig. 3a).19 Those peaks were slightly red-shifted compared with those of 1-ethynyl pyrene molecularly dissolved in DMF (Fig. S10, ESI†), suggesting that the fluorescence of pyrene units was influenced by the formation of covalent bonds with cellulose oligomers through the click reaction.
The pyrene-conjugated 2D crystalline cellulose oligomers dispersed in DMF showed broad fluorescence emission above 450 nm (Fig. 3a), which was different from that of reference 1-ethynyl pyrene (Fig. S11, ESI†). This observation suggested that certain amounts of pyrene units formed excimers on the nanomaterial surface. Although the ideal distance between pyrene units (at least 0.8 nm) on the nanomaterial surface is too large to form excimers (Fig. S12, ESI†),20 they were formed by partial contact of the 2D crystalline cellulose oligomers in DMF. The fluorescence emission derived from the excimers relatively increased with increasing concentration of pyrene-conjugated 2D crystalline cellulose oligomers (Fig. 3a), supporting the contribution of the concentration-dependent formation of the contacted pyrene units. When the pyrene-conjugated 2D crystalline cellulose oligomers were excited at 440 nm, which did not correspond to the absorption of pyrene monomers, broad fluorescence emission above 450 nm was observed (Fig. S13, ESI†), confirming the presence of static excimers,19,21 which were already formed in the ground states. More significantly, the circular dichroism (CD) spectra in DMF (0.034% (w/v)) showed a negative Cotton effect at 368 nm, which corresponded to the absorption of pyrene units (Fig. 3b and Fig. S9, ESI†). This observation suggested that the pyrene units existed in twisted arrangements between two 2D nanomaterials.22 It was therefore found that 2D crystalline cellulose oligomers have the potential to induce CD activities to covalently conjugated achiral dye molecules.
The fluorescence spectra of pyrene-conjugated 2D crystalline cellulose oligomers were measured in a variety of solvents and were different in each solvent (Fig. 4a). The monomer peaks were independent of the solvent species; however, the broad excimer peaks changed. Because the spectra of 1-ethynyl pyrene dissolved in the same solvents, except for water, were almost the same, independent of solvent species (Fig. S11, ESI†), the spectral difference was derived from the slight difference in the contacting states of pyrene units on the nanomaterial surface. Meaningfully, the color of the dispersions under UV light of 365 nm was dependent on the solvent species (Fig. 4b), demonstrating the solvatochromic properties. The color gradually changed from blue for toluene to green for water with increasing dielectric constant. These observations suggested that 2D crystalline cellulose oligomers behaved as novel platforms for controlling the excimer formation of conjugated dye molecules. To the best of our knowledge, this is the first demonstration of solvatochromism using dye-conjugated 2D organic nanomaterials.
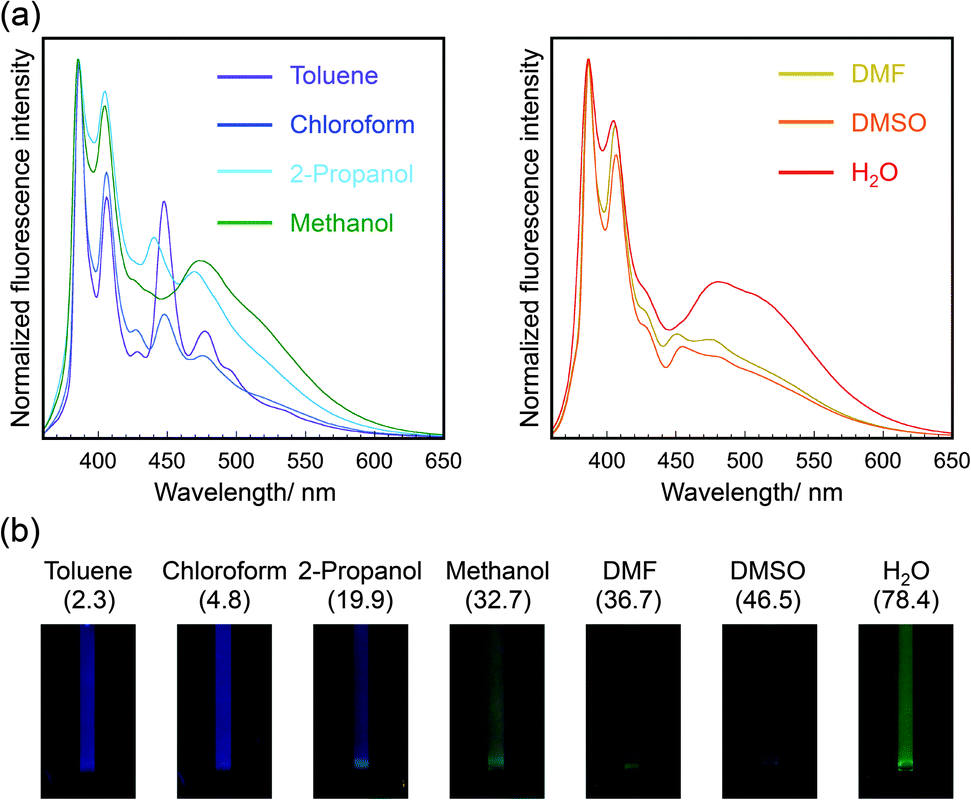 | ||
| Fig. 4 (a) Normalized fluorescence spectra (λex = 343 nm) of pyrene-conjugated 2D crystalline cellulose oligomers dispersed in seven solvents. The fluorescence intensity was normalized based on the intensity at 386 nm. The left and right graphs represent the fluorescence spectra dispersed in solvents with lower and higher dielectric constants, respectively. (b) Photographic images of pyrene-conjugated 2D crystalline cellulose oligomers dispersed in seven solvents under UV light (365 nm). The dielectric constant for each solvent is shown in parentheses. | ||
In conclusion, it was demonstrated that 2D crystalline cellulose oligomers with surface-reactive azide groups were synthesized by one-step CDP-catalyzed phosphorolytic reactions of αG1P monomers and β-glucosyl azide primers under aqueous and mild conditions and were post-functionalized with 1-ethynyl pyrene through copper(I)-catalyzed Huisgen cycloaddition reactions in DMF. The chemical, crystal, and morphological structures of the reactive and post-functionalized 2D crystalline cellulose oligomers were characterized by spectroscopy and microscopy. The fluorescence spectra revealed that the pyrene units conjugated on the nanomaterial surface partially formed excimers on the nanomaterial surface, followed by broad fluorescence emission, induced CD, and solvatochromism. Although post-functionalization of surface-azidized 2D crystalline cellulose oligomers with 1-ethynyl pyrene was performed as a model system, diverse combinations of other reactive primers and functional small molecules are applicable. We will develop such diverse and versatile 2D organic nanomaterials from the viewpoints of sustainability and biocompatibility.
The authors thank Prof. M. Wada (Kyoto Univ.) for CDP preparations, Prof. S. Nojima and Dr H. Marubayashi (Tokyo Tech) for WAXD measurements, and the Center for Advanced Materials Analysis (Tokyo Tech) for elementary analyses and TEM observations. This study was partially supported by the Funding Program for Next Generation World-Leading Researchers (NEXT Program) and Grants-in-Aids for Scientific Research (26288056 and 26620174) from the Japan Society for the Promotion of Science, and the collaborative research with JX Nippon Oil & Energy.
Notes and references
- A. K. Geim and K. S. Novoselv, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed; C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- R. Ma and T. Sasaki, Adv. Mater., 2010, 22, 5082–5104 CrossRef CAS PubMed.
- X. Zhuang, Y. Mai, D. Wu, F. Zhang and X. Feng, Adv. Mater., 2015, 27, 403–427 CrossRef CAS PubMed; S.-L. Cai, W.-G. Zhang, R. N. Zuckermann, Z.-T. Li, X. Zhao and Y. Liu, Adv. Mater., 2015 DOI:10.1002/adma.201500124.
- K. Baek, G. Yun, Y. Kim, D. Kim, R. Hota, I. Hwang, D. Xu, Y. H. Ko, G. H. Gu, J. H. Suh, C. G. Park, B. J. Sung and K. Kim, J. Am. Chem. Soc., 2013, 135, 6523–6528 CrossRef CAS PubMed; P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef PubMed; M. J. Kory, M. Wörle, T. Weber, P. Payamyar, S. W. v. d. Poll, J. Dshemuchadse, N. Trapp and D. A. Schlüter, Nat. Chem., 2014, 6, 779–784 CrossRef PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed; D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952–14955 CrossRef PubMed; S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef PubMed.
- K. T. Nam, S. A. Shelby, P. H. Choi, A. B. Marciel, R. Chen, L. Tan, T. K. Chu, R. A. Mesch, B.-C. Lee, M. D. Connolly, C. Kisielowski and R. N. Zuckermann, Nat. Mater., 2010, 9, 454–460 CrossRef CAS PubMed; Y. Kim, S. Shin, T. Kim, D. Lee, C. Seok and M. Lee, Angew. Chem., Int. Ed., 2013, 52, 6426–6429 CrossRef PubMed; Y. Zheng, H. Zhou, D. Liu, G. Floudas, M. Wagner, K. Koynov, M. Mezger, H. J. Butt and T. Ikeda, Angew. Chem., Int. Ed., 2013, 52, 4845–4848 CrossRef PubMed; K.-D. D. Zhang, J. Tian, D. Hanifi, Y. Zhang, A. C. Sue, T.-Y. Y. Zhou, L. Zhang, X. Zhao, Y. Liu and Z.-T. T. Li, J. Am. Chem. Soc., 2013, 135, 17913–17918 CrossRef PubMed; Z. M. Hudson, C. E. Boott, M. E. Robinson, P. A. Rupar, M. A. Winnik and I. Manners, Nat. Chem., 2014, 6, 893–898 CrossRef PubMed.
- Y. Habibi, L. Lucia and O. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS PubMed.
- T. Uryu, J. Yamanouchi, T. Kato, S. Higuchi and K. Matsuzaki, J. Am. Chem. Soc., 1983, 105, 6865–6871 CrossRef CAS; T. Nishimura and F. Nakatsubo, Carbohydr. Res., 1996, 294, 53–64 CrossRef; F. Nakatsubo, H. Kamitakahara and M. Hori, J. Am. Chem. Soc., 1996, 118, 1677–1681 CrossRef; T. Nishimura and F. Nakatsubo, Tetrahedron Lett., 1996, 37, 9215–9218 CrossRef.
- S. Kobayashi, S.-i. Shoda and H. Uyama, Adv. Polym. Sci., 1995, 121, 1–30 CrossRef CAS; J.-i. Kadokawa, Chem. Rev., 2011, 111, 4308–4345 CrossRef PubMed.
- S. Kobayashi, J. Sakamoto and S. Kimura, Prog. Polym. Sci., 2001, 26, 1525–1560 CrossRef CAS.
- K. Sheth and J. K. Alexander, J. Biol. Chem., 1969, 244, 457–464 Search PubMed; H. Nakai, M. Hachem, B. O. Petersen, Y. Westphal, K. Mannerstedt, M. J. Baumann, A. Dilokpimol, H. A. Schols, J. Duus and B. Svensson, Biochimie, 2010, 92, 1818–1826 CrossRef CAS PubMed; E. C. O'Neill and R. A. Field, Carbohydr. Res., 2015, 403, 23–37 CrossRef PubMed.
- M. Arai, K. Tanaka and T. Kawaguchi, J. Ferment. Bioeng., 1994, 77, 239–242 CrossRef CAS; A. K. Choudhury, M. Kitaoka and K. Hayashi, Eur. J. Org. Chem., 2003, 2462–2470 CrossRef PubMed; K. Shintate, M. Kitaoka, Y.-K. Kim and K. Hayashi, Carbohydr. Res., 2003, 338, 1981–1990 CrossRef; Y.-H. P. Zhang and L. R. Lynd, Appl. Microbiol. Biotechnol., 2006, 70, 123–129 CrossRef PubMed; G. H. Tran, T. Desmet, M. R. M. de Groeve and W. Soetaert, Biotechnol. Prog., 2011, 27, 326–332 CrossRef PubMed; H. G. Tran, T. Desmet, K. Saerens, H. Waegeman, S. Vandekerckhove, M. D'hooghe, I. V. Bogaert and W. Soetaert, Bioresour. Technol., 2012, 115, 84–87 CrossRef PubMed.
- E. Samain, C. Lancelon-Pin, F. Férigo, V. Moreau, H. Chanzy, A. Heyraud and H. Driguez, Carbohydr. Res., 1995, 271, 217–226 CrossRef CAS.
- M. Hiraishi, K. Igarashi, S. Kimura, M. Wada, M. Kitaoka and M. Samejima, Carbohydr. Res., 2009, 344, 2468–2473 CrossRef CAS PubMed.
- M. L. Nelson and R. T. O'Connor, J. Appl. Polym. Sci., 1964, 8, 1311–1324 CrossRef CAS PubMed.
- S. Califano and G. Abbondanza, J. Chem. Phys., 1963, 39, 1016–1023 CrossRef CAS PubMed.
- Q. Yang and X. Pan, J. Appl. Polym. Sci., 2010, 117, 3639–3644 CAS.
- H. J. Kim, J. Hong, A. Hong, S. Ham, J. H. Lee and J. S. Kim, Org. Lett., 2008, 10, 1963–1966 CrossRef CAS PubMed.
- M. Kolski, C. R. Arunkumar and K. S. Kim, J. Chem. Theory Comput., 2013, 9, 847–856 CrossRef.
- F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS.
- Y. Nagata, T. Nishikawa and M. Suginome, Chem. Commun., 2012, 48, 11193–11195 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, photographic images of reaction solutions, NMR, ATR-FTIR, UV-Vis absorption, and fluorescence spectra, WAXD diagrams, a CPK model of cellodecaose, TEM images, the crystal lattice of the cellulose II allomorph, elementary analysis data. See DOI: 10.1039/c5cc04378f |
| This journal is © The Royal Society of Chemistry 2015 |