
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalysts with carbon-centered activity for CO2 reduction with boranes†
Yanxin
Yang
,
Maotong
Xu
and
Datong
Song
*
Davenport Chemical Research Laboratories, Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: dsong@chem.utoronto.ca
First published on 11th June 2015
Abstract
We report two organocatalysts for CO2 hydroboration to methylborylethers, which upon hydrolysis can produce methanol. These organocatalysts feature carbon-centered reversible CO2 binding, broad borane scopes, and high catalytic activities.
The use of fossil fuels has caused a drastic increase of CO2 emissions over the past few decades.1 While the increase in CO2 level in the atmosphere raises serious environmental concerns, it also presents an opportunity for using CO2 as a sustainable C1 feedstock for chemical syntheses.2 Many promising methods have been developed for the conversion of CO2 to value-added chemicals, such as carbonates and derivatives, carboxylic acids and derivatives, formaldehyde, CO, alkanes, methylamines, and methanol.2,3 The catalytic reduction of CO2 to methanol is particularly interesting as it converts the combustion product back to a liquid fuel. Three general routes have been reported for the catalytic conversion of CO2 to methanol: hydrogenation,4,5 hydrosilylation,6 and hydroboration.7 A few transition metal7 and main group metal8 catalysts have been reported for the hydroboration of CO2 into methylborylether, which upon hydrolysis produces methanol. The metal-free phosphine–borane frustrated Lewis pairs (FLPs)9 and a few borohydride species10 have also shown catalytic activity towards the same transformation. Most of the above catalysts are plagued with intrinsic air- and moisture-sensitivity. In 2014, Cantat and co-workers reported the air- and moisture-stable N-heterocycle-based catalysts that only contain carbon, hydrogen, and nitrogen. These catalysts feature nitrogen-centered activity for catalysis.11 Unfortunately, these catalysts have limited borane scope, i.e., 9-borabicyclo[3.3.1]nonane (9-BBN) only. Despite these recent advances in CO2 reduction into methanol via catalytic hydroboration with heteroatom-centered reactivity, a catalyst with carbon-centered reactivity is unknown for this transformation. It is worth pointing out that although N-heterocyclic carbenes (NHCs) are known to catalyze the hydrosilylation of CO2 to methylsilylethers6a,b and the methylation of amines using CO2 as the carbon source,12 the hydroboration of CO2 into methylborylethers catalyzed by NHCs remains unknown.
Previously our group discovered the reversible CO2 insertion into the C–H bond of the actor 4,5-diazafluorenyl ligand supported by spectator metal centers.13 To confirm the spectator role of the metal centers, we further demonstrated this new reactivity with a metal-free compound, N-methyl-4,5-diazafluorenide, 1 (Scheme 1) by replacing spectator metal centers with a methyl group.13b To probe what structural features are essential for this new type of CO2 reactivity, we simplified the molecule from the three-ring system in 1 to a two-ring system in 2 (Scheme 1), because one of the pyridine moieties (color coated in gray) has no obvious role in CO2 binding.‡ Gratifyingly, 2 can indeed react with CO2 reversibly by inserting CO2 into the C–H bond (Scheme 1) on the C5 ring.‡ Both 1 and 2 bind with CO2 at the reactive carbon center, which is reminiscent of CO2-binding activity of NHCs.14 The carbon-centered CO2-binding property of 1 and 2 led us to explore the catalytic activity of these compounds toward hydroboration of CO2. The results are reported herein. To the best of our knowledge, the air- and moisture-stable and C/H/N-only compounds 1 and 2 are the first examples of carbon-centered catalysts for the hydroboration of CO2 into methylborylethers.
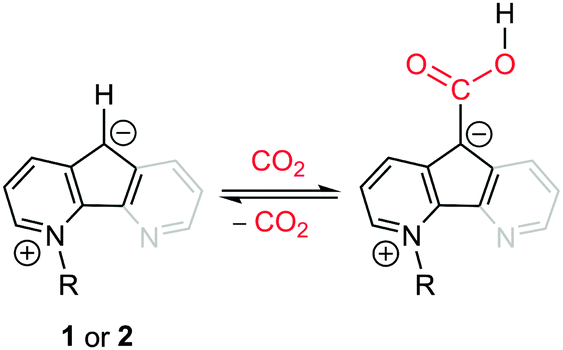 | ||
| Scheme 1 Reversible CO2 binding by 1 (R = Me, with the gray portion) and 2 (R = n-Pr, without the gray portion). | ||
Our initial tests showed that 1 could catalyze the hydroboration of CO2 with 9-BBN and catecholborane (HBcat). For example, when a C6D5Br solution of 1 and 10 eq. of 9-BBN was heated at 70 °C overnight under 1 atm of 13CO2, the major product 13CH3OBBN was observed along with a small amount of 13CH2(OBBN)2 in the 1H and 13C NMR spectra (see Fig. S16 and S17, ESI†). Similarly, when a C6D5Br solution of 1 and 30 eq. of HBcat was heated at 70 °C under 1 atm of 13CO2 for 2 h, NMR experiments showed that 13CH3OBcat was the only 13CO2 reduction product (see Fig. S18 and S19, ESI†). These preliminary results encouraged us to test the catalytic performance of 1 further. A C6D5Br solution of 1 and 100 eq. of HBcat was exposed to 1.5 atm of CO2 at 25 °C and the reaction was monitored with 1H and 11B NMR spectroscopy. The plot of TON vs. time for this reaction is shown in Fig. 1. The reaction started with a short induction period followed by fast catalysis. As HBcat was getting depleted toward the end of the reaction, the reaction rate was approaching 0. No induction period was observed at 70 °C and the reaction profile consisted of two stages: fast catalysis and plateau. The TOFs at the fast catalysis stage of the reactions were extracted from the plot: 41 and 231 h−1 for 25 °C and 70 °C reactions, respectively. Such TOFs put 1 amongst the most active organocatalysts for this transformation.
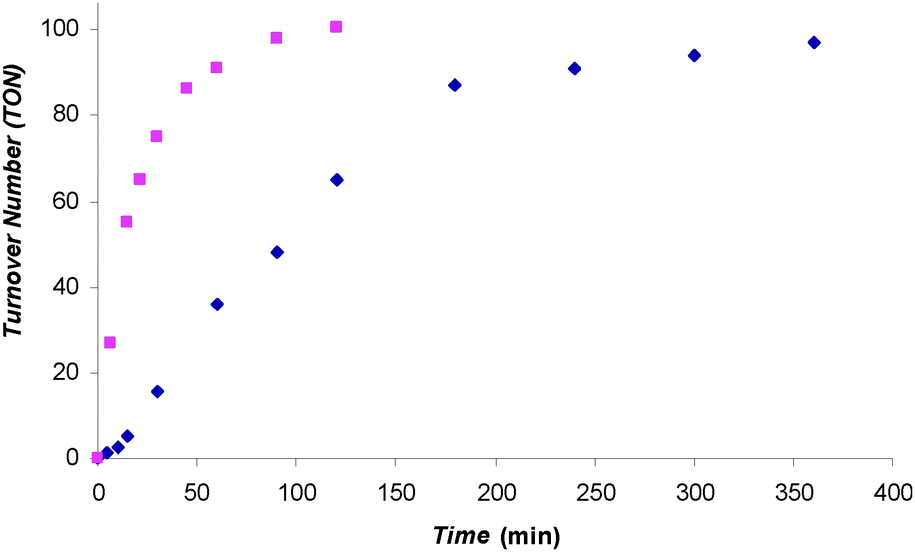 | ||
Fig. 1 TON vs. time plot for the formation of CH3OBcat catalyzed by 1. A C6D5Br solution of 1 and 100 eq. of HBcat was exposed to 1.5 atm CO2 at 70 °C ( ) and 25 °C ( ) and 25 °C ( ). ). | ||
When a C6D5Br solution of 1 and 100 eq. of HBcat was exposed to 1.5 atm of CO2 at 25 °C, CH3OBcat was produced with a TON of 97 within 6 h (Table 1, entry 1). When the same reaction was carried out at 70 °C, the reaction reached completion within 2 h (Table 1, entry 2); to test whether the catalyst was still active after 100 turnovers, 21.5 h after the complete consumption of the first 100 eq. of HBcat, another 100 eq. of HBcat was added to the reaction mixture, which was then re-charged with 1.5 atm of CO2 and reheated at 70 °C. The second batch of HBcat was consumed within 3 h to give an overall TON of 196 (Table 1, entry 3), indicating that catalyst 1 was still highly active.§
| Entry | Cat. | Borane | Solvent | T (°C) | Time (h) | TONb from the formation of each product | Total TONb | Avg. TOF (h−1) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HCO2BR2 | CH2(OBR2)2 | CH3OBR2 | (CH3OBO)3 | ||||||||
| a Reaction conditions: a Schlenk bomb was charged with 1.0 mg 1 or 2 and 100 eq. of borane, ∼0.6 mL of a deuterated solvent, and 1.5 atm¶ of CO2. The internal standard, hexamethylbenzene was added to the reaction mixture upon completion. b TON is based on the number of C–H bonds formed in the reduced products per molecule of the catalyst, determined by 1H NMR integration against the internal standard. c The number in parentheses is the TOF at the fast catalysis stage of the reaction. d The second 100 eq. of HBcat was added 21.5 h after the complete consumption of the first 100 eq. e The two numbers are the time required to consume the two batches of HBcat, respectively. f Control experiment (i.e., same conditions except for the absence of the catalyst) for entry 8 showed 4.5% conversion of BH3·SMe2 to (CH3OBO)3, while all other entries have no background reactions. | |||||||||||
| 1 | 1 | HBcat | C6D5Br | 25 | 6 | 97 | 97 | 16 (41)c | |||
| 2 | 1 | HBcat | C6D5Br | 70 | 2 | 100 | 100 | 50 (231)c | |||
| 3d | 1 | HBcat | C6D5Br | 70 | 2 + 3e | 196 | 196 | 39 | |||
| 4 | 1 | 9-BBN | C6D5Br | 25 | 8 | 11 | 47 | 58 | 7.3 | ||
| 5 | 1 | 9-BBN | C6D5Br | 70 | 2 | 16 | 51 | 67 | 34 | ||
| 6 | 1 | HBpin | CDCl3 | 100 | 48 | 6.82 | 0.53 | 6.28 | 13.6 | 0.28 | |
| 7 | 1 | BH3·SMe2 | C6D6 | 25 | 44 | 294 | 294 | 6.7 | |||
| 8 | 1 | BH3·SMe2 | CDCl3 | 70 | 20.5 | 286f | 286 | 14.0 | |||
| 9 | 2 | 9-BBN | CDCl3 | 25 | 19 | 12.5 | 48.6 | 61 | 3.2 | ||
| 10 | 2 | 9-BBN | CDCl3 | 70 | 2 | 7.2 | 59 | 66.2 | 33 | ||
| 11 | 2 | HBpin | CDCl3 | 90 | 46 | 1.6 | 73 | 74.6 | 1.6 | ||
| 12 | 2 | HBcat | CDCl3 | 25 | 19 | 97 | 97 | 5.1 | |||
| 13 | 2 | BH3·SMe2 | CDCl3 | 25 | 7 | 298 | 298 | 42.6 (55.6)c | |||
When 9-BBN was used as the reductant under the same conditions, the formations of CH2(OBBN)2 and CH3OBBN were observed at 25 °C within 8 h with a total TON of 58 (Table 1, entry 4); if the same reaction was carried out at 70 °C, the TON reached 66 within 2 h (Table 1, entry 5) and again CH2(OBBN)2 and CH3OBBN were both produced. The lower reaction rates in entries 4 and 5 compared to those in entries 1 and 2, respectively, could be attributed to the low solubility of 9-BBN. When a less reactive reductant pinacolborane (HBpin) was used, the catalytic reaction only gave 14 total turnovers in 48 h at 100 °C, yielding three reduction products HCOOBpin, CH2(OBpin)2 and CH3OBpin (Table 1, entry 6). Using 100 eq. of BH3·SMe2 (with respect to catalyst 1) as the reductant under 1.5 atm of CO2 the reaction achieved a TON of 294 with BH3 within 44 h at 25 °C to yield (CH3OBO)3 (Table 1, entry 7). Increasing the reaction temperature from 25 °C to 70 °C only improved the reaction rate by a factor of ∼2 (Table 1, entry 8).
Next, we tested the catalytic activity of 2. When a CDCl3 solution of 2 and 100 eq. of 9-BBN was exposed to 1.5 atm of CO2 at 25 °C, CH2(OBBN)2 and CH3OBBN were produced with an overall TON of 61 within 19 h (Table 1, entry 9); the reaction is much slower than that catalyzed by 1. When the same reaction was carried out at 70 °C, however, the reaction rate is comparable to that catalyzed by 1 at 70 °C, i.e., the reaction reached 66 TON within 2 h (Table 1, entry 10). Compared to 1, 2 showed a higher activity when HBpin was used as the reductant, i.e., the reaction catalyzed by 2 gave CH3OBpin as the dominant CO2 reduction product with a TON of 75 in 46 h at 90 °C (Table 1, entry 11). In contrast, when HBcat was used as the reductant, catalyst 2 showed lower activity than 1 (Table 1, entry 12). We speculate that the difference in catalytic activity between 1 and 2 may originate partly from the preferred interactions between the catalyst and borane: the larger π-system in 1 interacts with the aromatic backbone of HBcat more strongly, while the longer aliphatic propyl chain and smaller π-system in 2 favor the aliphatic backbone of HBpin. Interestingly, 2 showed much higher catalytic activity than 1 when BH3·SMe2 was used as the reductant, i.e., complete conversion to (CH3OBO)3 was observed in 7 h at 25 °C with a TON of 298 and average TOF of 42.6 h−1 (Table 1, entry 13). This reaction also has a short induction period at 25 °C (Fig. 2). A TOF of 56 h−1 at the fast catalysis stage was extracted from the plot of TON vs. time. Such TOFs make 2 one of the best organocatalysts for this transformation.
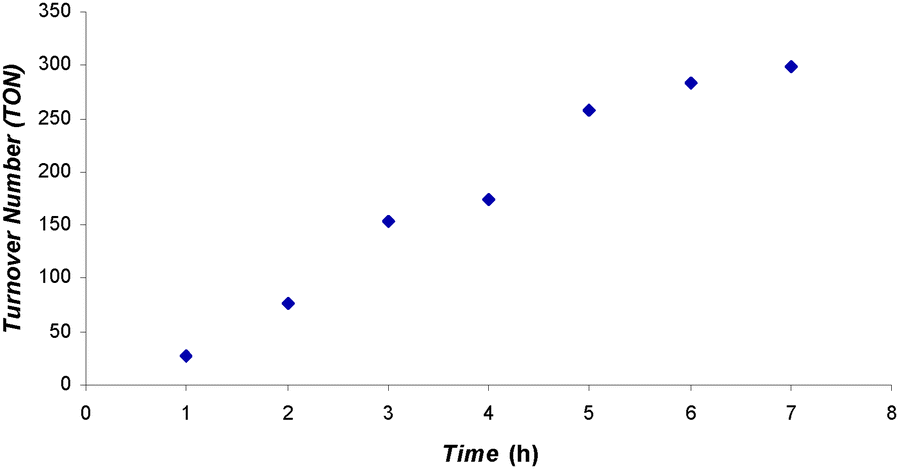 | ||
| Fig. 2 TON vs. time plot for the formation of (CH3OBO)3 catalyzed by 2. A CDCl3 solution of 2 and 100 eq. of BH3·SMe2 was exposed to 1.5 atm of CO2 at 25 °C. | ||
In summary, we have demonstrated compounds 1 and 2 not only bind CO2 reversibly via the formal insertion of CO2 into a C–H bond of the C5 ring, but also catalyze the hydroboration of CO2 to methylborylethers which upon hydrolysis can produce methanol. These air- and moisture-stable compounds that consist of only carbon, hydrogen, and nitrogen are the first catalysts with carbon-centered activity for the reduction of CO2 to methylborylethers. These catalysts feature broad borane scope and their catalytic activities are comparable to the best organocatalysts with heteroatom-based activity. The mechanism of the catalytic reactions are currently being investigated via experimental and computational methods in our laboratory.
We acknowledge Natural Science and Engineering Research Council of Canada (NSERC) for funding. Y. Y. greatly thanks the government of Ontario for an Ontario Trillium Scholarship. M. X. gratefully thanks University of Toronto for the University of Toronto Excellence Award and Charlie Kivi for X-ray crystallography.
Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010 Search PubMed.
- (a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed; (b) E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC; (c) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kuhn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed; (d) D. Yu, S. P. Teong and Y. Zhang, Coord. Chem. Rev., 2015, 293–294, 279–291 CrossRef CAS PubMed; (e) A. M. Appel, et al. , Chem. Rev., 2013, 133, 6621–6658 CrossRef PubMed; (f) D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625–2641 RSC; (g) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (h) C. S. Yeung and V. M. Dong, Top. Catal., 2014, 57, 1342–1350 CrossRef CAS PubMed; (i) G. Sahara and O. Ishitani, Inorg. Chem., 2015, 54, 5096–5104 CrossRef CAS PubMed.
- For CO2 hydrogenation to methanol, see recent reviews: (a) Y.-N. Li, R. Ma, L.-N. He and Z.-F. Daio, Catal. Sci. Technol., 2014, 4, 1498 RSC; (b) Y. Li, K. Junge and M. Beller, ChemCatChem, 2013, 5, 1072 CrossRef CAS PubMed; (c) E. Alberico and M. Nielsen, Chem. Commun., 2015, 51, 6714–6725 RSC and references therein.
- (a) N. M. Rezayee, C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS PubMed; (b) A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839–9843 CrossRef CAS PubMed.
- (a) S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS PubMed; (b) F. Huang, G. Lu, L. Zhao, H. Li and Z.-X. Wang, J. Am. Chem. Soc., 2010, 132, 12388 CrossRef CAS PubMed; (c) A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660–10661 CrossRef CAS PubMed; (d) S. Park, D. Bézier and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11404–11407 CrossRef CAS PubMed; (e) A. Berkefeld, W. E. Piers, M. Parvez, L. Castro, L. Maron and O. Eisenstein, Chem. Sci., 2013, 4, 2152–2162 RSC.
- (a) S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872–8873 CrossRef CAS PubMed; (b) F. Huang, C. Zhang, J. Jiang, Z.-X. Wang and H. Guan, Inorg. Chem., 2011, 50, 3816–3825 CrossRef CAS PubMed; (c) S. Chakraborty, Y. J. Patel, J. A. Krause and H. Guan, Polyhedron, 2012, 32, 30–34 CrossRef CAS PubMed; (d) S. Chakraborty, J. Zhang, Y. J. Patel, J. A. Krause and H. Guan, Inorg. Chem., 2013, 52, 37–47 CrossRef CAS PubMed; (e) M. J. Sgro and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 11343–11345 CrossRef CAS PubMed; (f) S. Bontemps, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 1671–1674 CrossRef CAS PubMed.
- (a) M. D. Anker, M. Arrowsmith, P. Bellham, M. S. Hill, G. Kociok-Kohn, D. J. Liptrot, M. F. Mahon and C. Weetman, Chem. Sci., 2014, 5, 2826–2830 RSC; (b) J. A. B. Abdalla, I. M. Riddlestone, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 5098–5012 CrossRef CAS PubMed.
- (a) M.-A. Courtemanche, M.-A. Légaré, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2013, 135, 9326–9329 CrossRef CAS PubMed; (b) M.-A. Légaré, M.-A. Courtemanche and F.-G. Fontaine, Chem. Commun., 2014, 50, 11362–11365 RSC; (c) M.-A. Courtemanche, M.-A. Legare, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2014, 136, 10708–10717 CrossRef CAS PubMed; (d) T. Wang and D. W. Stephan, Chem. Commun., 2014, 50, 7007–7010 RSC; (e) T. Wang and D. W. Stephan, Chem. – Eur. J., 2014, 20, 3036–3039 CrossRef CAS PubMed; (f) R. Declercq, G. Bouhadir, D. Bourissou, M.-A. Légaré, M.-A. Courtemanche, K. S. Nahi, N. Bouchard, F.-G. Fontaine and L. Maron, ACS Catal., 2015, 5, 2513–2520 CrossRef CAS.
- (a) S. Y.-F. Ho, C.-W. So, N. S. Merceron and N. Mézailles, Chem. Commun., 2015, 51, 2107–2110 RSC; (b) K. Fujiwara, S. Yasuda and T. Mizuta, Organometallics, 2014, 33, 6692–6695 CrossRef CAS; (c) Z. Lu, H. Hausmann, S. Becker and H. A. Wegner, J. Am. Chem. Soc., 2015, 137, 5332–5335 CrossRef CAS PubMed; (d) I. Knopf and C. C. Cummins, Organometallics, 2015, 34, 1601–1603 CrossRef CAS.
- C. Das Neves Gomes, E. Blondiaux, P. Thuéry and T. Cantat, Chem. – Eur. J., 2014, 23, 7098–7106 CrossRef PubMed.
- E. Blondiaux, J. Pouessel and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 12186–12190 CrossRef CAS PubMed.
- (a) V. T. Annibale and D. Song, Chem. Commun., 2012, 48, 5416–5418 RSC; (b) V. T. Annibale and D. Song, J. Am. Chem. Soc., 2013, 135, 16175–16183 CrossRef CAS PubMed.
- H. A. Duong, T. N. Tekavec, A. M. Arif and J. Louie, Chem. Commun., 2004, 112–113 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details. CCDC 1400031. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc04337a |
| ‡ Compound 2 is an orange oil and can be synthesized using a modified literature procedure. Compared to 1, whose solution is stable in air for several hours, the solution of 2 can be stored at −15 °C in air for weeks without significant change. Compound 2 is soluble in all common organic solvents. For the synthetic protocol of 2 and CO2 binding experiments, see ESI.† |
| § The slightly slower conversion of the second batch of HBcat was likely due to the inefficient mixing of the reactants caused by the large amount of solid produced in the reaction. |
| ¶ The Schlenk bomb charged with all other reagents and solvents was immersed in liquid N2 to freeze the solution; the headspace was then evacuated. The entire bomb was then immersed in a −70 °C dry ice-isopropanol bath to keep the solution frozen and cool the headspace. The bomb was then opened to 1 atm of CO2 for 10 minutes to allow the temperature to equilibrate. Subsequently the bomb was sealed and allowed to warm to 25 °C to achieve ∼1.5 atm pressure. Safety warning: if CO2 gas was introduced below −78 °C, dry ice would condense in the reaction vessel and the final pressure becomes time-dependent and can no longer be calculated easily. Using our protocol with a low-melting solvent (i.e., the solvent is not frozen at −70 °C), the final pressure is again time-dependent, because of the dramatically increased solubility of CO2 at −70 °C. In both scenarios prolonged CO2 exposure could cause serious explosions due to uncontrolled high pressures and make the results incomparable to others due to unknown CO2 pressure. |
| This journal is © The Royal Society of Chemistry 2015 |