
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile rotation around a silicon–phosphorus double bond enabled through coordination to tungsten†
Nora C.
Breit
a,
Tibor
Szilvási
b and
Shigeyoshi
Inoue
*a
aInstitut für Chemie, Technische Universität Berlin, Straße des 17. Juni 135, Sekr. C2, D-10623 Berlin, Germany. E-mail: shigeyoshi.inoue@tu-berlin.de
bDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Szent Gellért tér 4, 1111 Budapest, Hungary
First published on 10th June 2015
Abstract
Unprecedented E/Z isomerisation of a Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P bond was observed by temperature dependent NMR spectroscopy. DFT calculations showed that the coordination of phosphasilene to tungsten lowered the rotational barrier from 19.1 to 14.2 kcal mol−1. The thermodynamically more stable phosphinosilylene tungsten complex is formed at elevated temperatures through substituent migration.
P bond was observed by temperature dependent NMR spectroscopy. DFT calculations showed that the coordination of phosphasilene to tungsten lowered the rotational barrier from 19.1 to 14.2 kcal mol−1. The thermodynamically more stable phosphinosilylene tungsten complex is formed at elevated temperatures through substituent migration.
It is well known that a π bond is usually not free to rotate. For instance, alkenes have very high activation energies (cis-2-butene = 62 kcal mol−1) for rotation around the C
C bond, making them inert against E/Z-isomerisation at room temperature.1 However, this isomerisation energy can be decreased by introducing bulky substituents (cf. stilbene = 43 kcal mol−1, bifluorenylidene = 23 kcal mol−1) or using the push–pull effect1c (cf. dimethyl[(dimethylamino)methylene]malonate = 16 kcal mol−1).
Their higher homologues, disilenes, display relatively low rotation barriers of 26–28 kcal mol−1 at 350 K for the SiSi bond, which enable E/Z-isomerisation.2 In the interesting case of tetrakis(trialkylsilyl)-disilenes, rotation around the SiSi bond becomes possible at room temperature (activation barrier 15.3 kcal mol−1 at 303 K), which is attributed to an effective σ–π conjugation in the transition state.3 It should be noted that additional pathways for E/Z-isomerisation of disilenes through dissociation–recombination reactions, disilene–silylsilylene interconversions and through irradiation are also viable.4 Diphosphenes with a PP double bond can undergo photoisomerisation from E- to Z-conformations.5 However, to the best of our knowledge no E/Z-isomerisation was reported for phosphasilenes (Chart 1). The SiP bond can be strongly influenced by the introduction of substituents that can enhance the elemental polarization induced by the different electronegativities of silicon and phosphorus.6 The push–pull effect could also be used for decreasing the rotation barrier of the SiP double bond with the intention to observe E/Z-isomerisation of phosphasilenes. In addition, phosphasilenes feature an electron lone pair on phosphorus that can coordinate to transition metals. This coordination behaviour on phosphorus may lead to a lower rotation energy of the SiP double bond. However, surprisingly, little is known about its coordination to transition metals.7
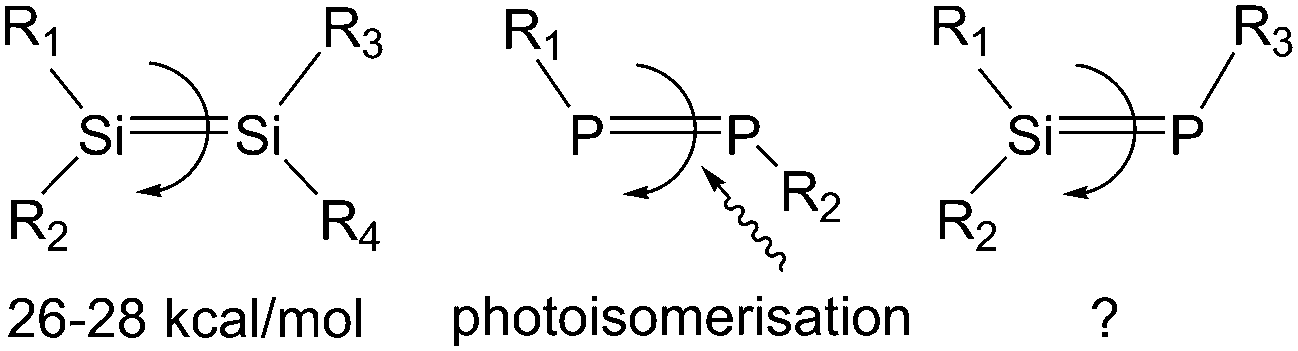 | ||
| Chart 1 E/Z-Isomerisation of compounds containing a double bond. | ||
With this in mind, we utilized zwitterionic phosphasilene 16a and engaged the electron lone pair on phosphorus in coordination to tungsten (Scheme 1). By this we obtained an even weaker SiP bond in 2 and a low rotation barrier of only 14.2 kcal mol−1 leading to fast E/Z-isomerisation at room temperature.
 | ||
| Scheme 1 Synthesis and isomerism of complex 2. | ||
The phosphasilene tungsten carbonyl complex 2 was synthesized from 1 and W(CO)5·thf in a very good yield of 92% (Scheme 1). Compound 2 exists as two stable isomers labeled as (E)-2 and (Z)-2. It should be noted that only (E)-2 is having the proper Si2–Si1–P1–W1 torsion angle of 175.13°, while the isomer referred to as (Z)-2 actually exhibits a Si2–Si1–P1–W1 torsion angle of 38.8°. The rotational isomer with a torsion angle of 0° is higher in energy (6.8 kcal mol−1, see the ESI,† Fig. S24). DFT calculations based on the B97-D/6-31G(d) level of theory revealed that the rotation barrier for the silicon–phosphorus double bond in 2 is 14.2 kcal mol−1, which is feasible at room temperature, but not at low temperature. Indeed, we observed by NMR spectroscopy that the signals belonging to one set at room temperature undergo coalescence and give rise to two sets of signals at −60 °C (Fig. 1). The ratio between (E)-2 and (Z)-2 is 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in favor of (E)-2, which was assigned based on lower thermodynamic stability of (Z)-2 (2.9 kcal mol−1). The most prominent differences in the NMR shifts of (E)-2 and (Z)-2 were observed for the phosphorus and the silene silicon. The 31P{1H} NMR signals of 2 ((E)-2: δ = −282.0 ppm, 1JP–W = 123 Hz and (Z)-2: δ = −304.3 ppm, 1JP–W = 128 Hz) are upfield shifted from that of 1 (δ = −252.9 ppm).6a The low tungsten–phosphorus coupling constants of 123 and 128 Hz are indicative of the strong negative polarization on phosphorus and can be compared to those of (phosphoranylidenephosphine)pentacarbonyltungsten complexes (102.5–109.9 Hz).8 The silene 29Si{1H} NMR signals of 2 ((E)-2: δ = 49.8 (1JSi–P = 106 Hz) ppm and (Z)-2: δ = 45.9 (1JSi–P = 130 Hz) ppm) are downfield shifted from that of 1 (δ = 40.5 (1JSi–P = 191.4 Hz) ppm).6a In addition the silene–phosphorus coupling constant is significantly decreased in 2 compared to 1, which is all in agreement with the weakened and more polarized SiP bond in 2. It is important to note that the NMR signals for the silene silicon and phosphorus are broad at room temperature presumably due to the rotation around this bond. Solid state 29Si{1H} and 31P{1H} NMR spectroscopy revealed that only (E)-2 is present in the solid state. Variable temperature 1H and 31P{1H} NMR measurements of phosphasilene 1 did not reveal coalescence or two sets of signals at low temperatures down to −60 °C, proving experimentally that a rotation around the SiP bond at r.t. is not applicable for 1.
:1 in favor of (E)-2, which was assigned based on lower thermodynamic stability of (Z)-2 (2.9 kcal mol−1). The most prominent differences in the NMR shifts of (E)-2 and (Z)-2 were observed for the phosphorus and the silene silicon. The 31P{1H} NMR signals of 2 ((E)-2: δ = −282.0 ppm, 1JP–W = 123 Hz and (Z)-2: δ = −304.3 ppm, 1JP–W = 128 Hz) are upfield shifted from that of 1 (δ = −252.9 ppm).6a The low tungsten–phosphorus coupling constants of 123 and 128 Hz are indicative of the strong negative polarization on phosphorus and can be compared to those of (phosphoranylidenephosphine)pentacarbonyltungsten complexes (102.5–109.9 Hz).8 The silene 29Si{1H} NMR signals of 2 ((E)-2: δ = 49.8 (1JSi–P = 106 Hz) ppm and (Z)-2: δ = 45.9 (1JSi–P = 130 Hz) ppm) are downfield shifted from that of 1 (δ = 40.5 (1JSi–P = 191.4 Hz) ppm).6a In addition the silene–phosphorus coupling constant is significantly decreased in 2 compared to 1, which is all in agreement with the weakened and more polarized SiP bond in 2. It is important to note that the NMR signals for the silene silicon and phosphorus are broad at room temperature presumably due to the rotation around this bond. Solid state 29Si{1H} and 31P{1H} NMR spectroscopy revealed that only (E)-2 is present in the solid state. Variable temperature 1H and 31P{1H} NMR measurements of phosphasilene 1 did not reveal coalescence or two sets of signals at low temperatures down to −60 °C, proving experimentally that a rotation around the SiP bond at r.t. is not applicable for 1.
 | ||
| Fig. 1 The dynamic behavior of 2 observed by temperature dependent NMR spectroscopy in toluene-d8. | ||
X-ray diffraction analysis of single crystals obtained from hexane elucidated the molecular structure of (E)-2 (Fig. 2). The Si1–P1 bond length (2.158(2) Å) in (E)-2 is clearly longer than that in 1 (2.095(3) Å),6a which confirms further the weakening of the silicon–phosphorus double bond by the coordination of phosphorus to tungsten. The coordination to gold in the only known phosphasilene complexes with a coordinative bond showed little effect on the Si1–P1 bond lengths (2.0924(2) Å and 2.0917(5) Å).7c The angles around phosphorus are nearly 120° and the torsion angle Si2–Si1–P1–W1 amounts to 175.13° establishing the η1-coordination of the SiP bond to tungsten. The P1–W1 bond length (2.6265(18) Å) is longer than in phosphaalkene tungsten complexes (2.51–2.54 Å)9 and closer to that of the (phosphoranylidenephosphine)penta-carbonyltungsten complex (2.603(1) Å).8b DFT calculations were utilized to understand the molecular structure of (Z)-2. Only minor differences were found for the interatomic distances in (Z)-2 and (E)-2 (Si1–P1 2.159 Å and 2.152 Å and P1–W1 2.663 Å and 2.648 Å, respectively).
 | ||
| Fig. 2 Molecular structure of compound (E)-2. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) in (E)-2: Si1–P1 2.158(2), Si1–Si2 2.369(2), P1–Si3 2.253(2), P1–W1 2.6265(18); Si1–P1–W1 118.82(8), Si3–P1–W1 115.55(8). | ||
UV/Vis spectroscopy revealed a red-shift from 1 to 2 (λmax = 333 nm and λmax = 354 nm, respectively), which was also observed for the phosphasilene gold complexes (8–10 nm).7c TD-DFT calculations based on the B3LYP/cc-pVTZ//B97-D/6-31G(d) level of theory similarly predicted a red shift for (E)-2 (λmax = 383 nm, 3.24 eV, oscillator strength 0.050). The UV/Vis transition of (E)-2 occurs mainly from the HOMO to the LUMO (weight 0.55), while the LUMO+1 and LUMO+2 (weight 0.11 and weight 0.15, respectively) play a less significant part (see the ESI,† Fig. S25). The HOMO of (E)-2 (−4.13 eV) is determined by the antibonding combination of the π orbital of the phosphasilene and a filled d-orbital of tungsten. It is higher in energy than the HOMO of 1 (−5.94 eV) marked by the silicon–phosphorus π-bonding orbital which can explain the observed red-shift.6e,10 According to NBO analysis, the already very polarized double bond of 1 (silicon: +0.99 charge, 21.45% of the π-bond, phosphorus: −0.75 charge, 78.55%)6e gets even further polarized in 2 (silicon: +1.14 charge, phosphorus: −0.49 charge), to the extent of being dissected by NBO analysis into an empty orbital on silicon and a lone pair on phosphorus.11 This confirms the increasing importance of zwitterionic resonance structures, also supported by the amidinato ligand and its additional coordination (Scheme 2), which explains the tendency of 2 for E/Z-isomerisation.
 | ||
| Scheme 2 Resonance structures of 2. | ||
Though compound 2 is stable at room temperature, at elevated temperatures it undergoes a slow reaction to yield the phosphinosilylene tungsten carbonyl complex 3 (Scheme 3).12 DFT calculations showed that a stepwise mechanism in contrast to a concerted shift of the TMS group and W(CO)5 moiety occurs for the transformation of 2 to 3 (see the ESI,† Fig. S26). The first step is the shift of the TMS group, which is also rate determining (transition state: 19.7 kcal mol−1) and thus responsible for the kinetic stability of 2 at room temperature. The formation of 3 from 2 is thermodynamically favoured by 10.1 kcal mol−1 and can be explained by the stronger donor strength of the silylene.13 In accordance with the calculated values, no formation of 2 from 3 at elevated temperatures was observed. This is contrasted by the equilibrium between 1 and phosphinosilylene 4 (Scheme 3), which is determined by a smaller thermodynamic difference (1.9 kcal mol−1 in favour of 1) and a higher activation barrier (32.1 kcal mol−1).14
 | ||
| Scheme 3 Reactivity of 2 and independent synthesis of 3 from 4. | ||
To date, merely four stable phosphinosilylenes have been reported15 and their reactivity studies were limited to N2O, tBuCOCl and Ni(COD)2.6d,14,16 Complex 3 was synthesized independently from the parent phosphinosilylene 4 in a moderate to low yield of 30% (Scheme 3).17 The silylene tungsten complex 3 was thus fully characterized using NMR and IR spectroscopy, mass spectrometry, elemental analysis and single crystal X-ray diffraction. As expected, the 29Si{1H} NMR signal of the silylene–silicon in 3 is downfield shifted from that of 4 and the 1JSi–P coupling constant has considerably decreased (δ = 70.7 ppm, 1JSi–P = 134 Hz and δ = 44.0 ppm, 1JSi–P = 194 Hz, respectively).6a The 31P{1H} NMR signal of 3 is slightly downfield shifted from that of 4 (δ = −199.4 ppm and δ = −211.0 ppm, respectively).6a The presence of the carbonyl group is evident from 13C{1H} NMR spectroscopy (δ = 202.0 ppm) and IR spectroscopy (νCO = 2054, 1918, 1905 cm−1). The molecular structure of compound 3 is shown in Fig. 3 and it revealed that the silicon–phosphorus bond length in 3 (2.249(9) Å and 2.237(6) Å) is shorter than that of 4 (2.2838(12) Å).6a The silicon–tungsten bond length in 3 (2.562(3) Å and 2.619(2) Å) is longer than those of (PhC{NtBu}2)Si(W{CO}5)Cl and (PhC{NtBu}2)Si(W{CO}5)F (2.5086(11) Å and 2.4990(8) Å, respectively)18 and similar to that of (PhC{NiPr}2)2Si(W{CO}5) (2.5803(9) Å).19
 | ||
| Fig. 3 Molecular structure of compound 3. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted and only one of the disordered fractions is depicted for clarity. Selected bond lengths (Å) and angles (°) in 3, values in brackets refer to the other disordered fraction: Si1–P1 2.249(9) {2.237(6)}, Si1–W1 2.562(3) {2.619(2)}, P1–Si2 2.228(13) {2.260(10)}, P1–Si3 2.177(10) {2.375(9)}, W1–Si1–P1 122.0(3) {120.7(2)}. | ||
In conclusion, the coordination of zwitterionic phosphasilene 1 to a tungsten carbonyl complex leads to a more polarized and weaker silicon–phosphorus bond in 2. Interestingly, it also lowers the rotation barrier of the SiP bond from 19.1 kcal mol−1 in 1 to 14.2 kcal mol−1 in 2 to allow facile E/Z-isomerisation at room temperature. In the presence of the transition metal, the phosphinosilylene complex 3 is thermodynamically preferred to 2. Both species, 2 and 3, are versatile building blocks for low-valent silicon compounds due to their labile trimethylsilyl groups6a,e,14 and the additional stabilization provided by the transition metal. In fact, further reactivity studies are currently under way and shall be reported in due course.
Notes and references
- (a) E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, John Wiley & Sons, Ltd, New York, 1994, pp. 544–550 Search PubMed; (b) I. Fleming, Molecular Orbitals and Organic Chemical Reactions, John Wiley & Sons, Ltd, 2010, p. 101 Search PubMed; (c) Y. Shvo, E. C. Taylor and J. Bartulin, Tetrahedron Lett., 1967, 3259 CrossRef CAS.
- Selected observations and calculations for thermodynamic E/Z-isomerisation via rotation of SiSi bonds:
(a) M. J. Michalczyk, R. West and J. Michl, Organometallics, 1985, 4, 826 CrossRef CAS;
(b) S. A. Batcheller, T. Tsumuraya, O. Tempkin, W. M. Davis and S. Masamune, J. Am. Chem. Soc., 1990, 112, 9394 CrossRef CAS.
- M. Kira, S. Ohya, T. Iwamoto, M. Ichinohe and C. Kabuto, Organometallics, 2000, 19, 1817 CrossRef CAS.
- First example of thermal dissociation of a disilene into a silylene: (a) N. Tokitoh, H. Suzuki and R. Okazaki, J. Am. Chem. Soc., 1993, 115, 10428 CrossRef CAS . Example for a dyotropic rearrangement rather than a disilene–silylsilylene interconversion: ; (b) H. B. Yokelson, J. Maxka, D. A. Siegel and R. West, J. Am. Chem. Soc., 1986, 108, 4239 CrossRef CAS . For a review see: ; (c) M. Kira, Proc. Jpn. Acad., Ser. B, 2012, 88, 167 CrossRef CAS.
- Selected examples: (a) A.-M. Caminade, M. Verrier, C. Ades, N. Paillous and M. Koenig, J. Chem. Soc., Chem. Commun., 1984, 875 RSC; (b) M. Yoshifuji, T. Hashida, N. Inamoto, K. Hirotsu, T. Horiuchi, T. Higuchi, K. Ito and S. Nagase, Angew. Chem., Int. Ed. Engl., 1985, 24, 211 CrossRef; (c) E. Niecke, O. Altmeyer and M. Nieger, Angew. Chem., Int. Ed. Engl., 1991, 30, 1136 CrossRef.
- Selected zwitterionic phosphasilenes: (a) S. Inoue, W. Wang, C. Präsang, M. Asay, E. Irran and M. Driess, J. Am. Chem. Soc., 2011, 133, 2868 CrossRef CAS PubMed; (b) S. S. Sen, S. Khan, H. W. Roesky, D. Kratzert, K. Meindl, J. Henn, D. Stalke, J.-P. Demers and A. Lange, Angew. Chem., Int. Ed., 2011, 50, 2322 CrossRef CAS PubMed; (c) K. Hansen, T. Szilvási, B. Blom, S. Inoue, J. Epping and M. Driess, J. Am. Chem. Soc., 2013, 135, 11795 CrossRef CAS PubMed; (d) K. Hansen, T. Szilvási, B. Blom, E. Irran and M. Driess, Chem. – Eur. J., 2014, 20, 1947 CrossRef CAS PubMed; (e) N. C. Breit, T. Szilvási and S. Inoue, Chem. – Eur. J., 2014, 20, 9312 CrossRef CAS PubMed.
- One phosphasilene iron complex was observed using 29Si{1H} and 31P{1H} NMR spectroscopy and mass spectrometry. (a) M. Driess, H. Pritzkow and U. Winkler, J. Organomet. Chem., 1997, 529, 313 CrossRef CAS. One phosphasilene zinc complex with a mainly covalent bond that remains in (E)-configuration at variable temperatures was reported. (b) M. Driess, S. Block, M. Brym and M. T. Gamer, Angew. Chem., Int. Ed., 2006, 45, 2293 CrossRef CAS PubMed. Few phosphasilene gold complexes with a coordination of the lone pair on phosphorus were described. (c) B. Li, T. Matsuo, T. Fukunaga, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, Organometallics, 2011, 30, 3453 CrossRef CAS.
- (a) F. Mercier, F. Mathey, C. Afiong-Akpan and J. F. Nixon, J. Organomet. Chem., 1988, 348, 361 CrossRef CAS; (b) P. Le Floch, A. Marinetti, L. Ricard and F. Mathey, J. Am. Chem. Soc., 1990, 112, 2407 CrossRef CAS.
- L. Weber, M. Meyer, H.-G. Stammler and B. Neumann, Chem. – Eur. J., 2001, 7, 5401 CrossRef CAS.
- For a more detailed description of the MOs of (E)-2 and 1, please see the ESI†.
- It is important to note that the increased positive charge on silicon represents the stronger polarization, since only silicon is in the same environment in 1 and 2, while the charge on phosphorus is significantly influenced by its coordination to tungsten.
- A flame sealed NMR tube containing a solution of 2 in C6D6 was heated to 70 °C, after a few hours the first signs of the formation of 3 were observed, but complete consumption of 2 took 7 weeks at 70 °C.
- Z. Benedek and T. Szilvási, RSC Adv., 2015, 5, 5077 RSC.
- N. C. Breit, T. Szilvási, T. Suzuki, D. Gallego and S. Inoue, J. Am. Chem. Soc., 2013, 135, 17958 CrossRef CAS PubMed.
- Stable phosphinosilylenes: (a) H. H. Karsch, U. Keller, S. Gamper and G. Müller, Angew. Chem., Int. Ed. Engl., 1990, 29, 295 CrossRef; (b) C.-W. So, H. W. Roesky, P. M. Gurubasavaraj, R. B. Oswald, M. T. Gamer, P. G. Jones and S. Blaurock, J. Am. Chem. Soc., 2007, 129, 12049 CrossRef CAS PubMed; (c) R. Azhakar, R. S. Ghadwal, H. W. Roesky, H. Wolf and D. Stalke, Organometallics, 2012, 31, 4588 CrossRef CAS , and 6a. Examples of transient phosphinosilylenes: ; (d) H. Cui, J. Zhang and C. Cui, Organometallics, 2013, 32, 1 CrossRef CAS; (e) D. Geiß, M. I. Arz, M. Straßmann, G. Schnakenburg and A. C. Filippou, Angew. Chem., Int. Ed., 2015, 54, 2739 CrossRef PubMed.
- R. Azhakar, K. Pröpper, B. Dittrich and H. W. Roesky, Organometallics, 2012, 31, 7586 CrossRef CAS.
- 3 is the main product in this reaction, but either W(CO)5·thf or 3 is catalyzing the formation of 1 from 4, so that 2 and sometimes 1 are observed as side products.
- R. Azhakar, R. S. Ghadwal, H. W. Roesky, H. Wolf and D. Stalke, J. Am. Chem. Soc., 2012, 134, 2423 CrossRef CAS PubMed.
- K. Junold, J. A. Baus, C. Burschka and R. Tacke, Angew. Chem., Int. Ed., 2012, 51, 7020 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures and characterization methods, NMR spectra, IR spectra, the UV/Vis spectrum, crystallographic and computational details, molecular orbitals and mechanistic schemes. CCDC 1061059 (2) and 1061060 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc04247j |
| This journal is © The Royal Society of Chemistry 2015 |