
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePulsed EPR spectroscopy distance measurements of DNA internally labelled with Gd3+-DOTA†
Filip
Wojciechowski
a,
Andreas
Groß
ab,
Isabelle T.
Holder
ab,
Laura
Knörr
a,
Malte
Drescher
*ab and
Jörg S.
Hartig
*ab
aDepartment of Chemistry, University of Konstanz, Universitätsstr. 10, Konstanz, Germany. E-mail: joerg.hartig@uni-konstanz.de; malte.drescher@uni-konstanz.de; Tel: +49-7531884575
bKonstanz Research School Chemical Biology, University of Konstanz, Germany
First published on 22nd July 2015
Abstract
Gd3+ is increasingly used in EPR spectroscopy due to its increased intracellular stability and signal-to-noise ratios. Here we present the incorporation of Gd3+-DOTA into internal positions in DNA. Distance measurements via pulsed Electron Paramagnetic Resonance (EPR) spectroscopy in vitro and in cellula proved enhanced stability and efficiency compared to nitroxide labels.
Pulsed electron paramagnetic resonance (EPR) spectroscopy has emerged as a powerful technique in the study of the molecular structure and dynamics of biomacromolecules under in vitro conditions1–3 as well as in the cellular milieu.4–10 The pulsed EPR method double electron–electron resonance (DEER) in combination with site-directed labelling (SDSL) of biomacromolecules facilitates distance measurements between two spin labels in the nanometer range.11–13 Typically, a paramagnetic label based on a stable nitroxide radical with the general structural building block of RNO˙ is used as a spin label. However, alternative paramagnetic compounds are being investigated for their use as EPR probes in biomacromolecules.
Within the context of oligonucleotides (DNA, RNA and non-canonical oligonucleotide structures) EPR spectroscopy has been proven to be extremely useful for measuring distance distributions and studying conformational changes in polymorphic non-canonical DNA structures called G-quadruplexes, which were labelled with SDSL.14–16 Similarly, EPR has been used to detect the formation of both duplex and triplex DNA structures.17 Recently, X-band continuous wave-EPR spectroscopy has been applied to detect abasic sites in duplex DNA as well as in base-pair mismatch detection.18
However, while nitroxide radicals have been extensively used in EPR spectroscopy in vitro, they have some drawbacks in the context of high field EPR measurements in cellula. When used in cell studies, the nitroxide radicals encounter the reducing environment of the cellular milieu which can quench the nitroxide to an EPR-silent N-hydroxyl derivative.6,7 To overcome this limitation and benefit from further advantages, especially gadolinium-based spin labels become more and more the focus of attention and were used in several approaches for pulsed EPR distance measurements.19–23 The lanthanide gadolinium in the oxidation state of 3+ has recently been used as a paramagnetic centre in in-cell EPR spectroscopy.9,10 Besides the stability of these spin labels inside the cellular milieu, they furthermore excel by no orientation selection, higher transition moments owing to the high spin system of S = 7/2, fast repetition rates due to the short longitudinal relaxation (T1) rates and therefore increased signal-to-noise-ratios of DEER experiments in comparison to standard nitroxide labels.19
The macrocycle 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) has been extensively used to chelate Gd3+.24–26 DOTA is one of the most powerful metal chelating agents for lanthanide ions27 and is used as a contrast agent in magnetic resonance imaging in the medical diagnosis.28,29 Recently Gd3+-DOTA has been attached by SDSL at the 5′-position of oligonucleotides using click chemistry and was used in model EPR distance measurements.30 However, future applications of Gd3+–Gd3+ DEER distance measurements in oligonucleotides would benefit from being able to internally label oligonucleotides at any desired position with Gd3+-DOTA. This would allow for studying a much broader range of nucleic acid structures via Gd3+–Gd3+ DEER measurements.
There are two main strategies for the internal labelling of oligonucleotides with Gd3+-DOTA; addition of a Gd3+-DOTA phosphoramidite during solid-phase synthesis or post-synthetic labelling either in solution or on the solid-phase resin. The synthesis of a DOTA phosphoramidite has been described.31 However, the DOTA unit was appended at the N3 position of uracil, thereby preventing the modified nucleobase to undergo Watson–Crick base-pairing and limiting its incorporation to the ends of oligonucleotides where it has a minimal destabilizing effect. Furthermore, due to the likely incompatibility of metalated DOTA (e.g. Gd3+-DOTA) in solid-phase oligonucleotide synthesis the metal needs to be introduced post-synthetically.
Therefore we have decided to pursue a post-synthetic labelling strategy on the solid-phase resin using copper-catalysed 1,3-dipolar cycloaddition reaction (click chemistry).32,33 There are many examples of click chemistry between alkyne modified oligonucleotides and azides.34 However the synthesis of azide labelled oligonucleotides is less common.35 This is due to the instability of azides to P(III) oligonucleotide chemistry: the azide undergoes Staudinger reaction during oligonucleotide synthesis.36
Herein, we describe the synthesis of a phosphoramidite building block that was used in the automated DNA synthesis of azides containing DNA, which was subsequently reacted with alkyne-(Gd3+-DOTA)37,38 to give internally labelled (Gd3+-DOTA)-DNA. We characterize a DNA duplex with respect to the influence of the Gd-DOTA label on the stability and structure of the dsDNA helix. Then, we performed EPR spectroscopy-based distance measurements of these modified oligonucleotides both in vitro and in cellula.
The C5 position of uracil was chosen to attach Gd3+-DOTA to DNA. Bulky substituents can be incorporated at this position without influencing the syn/anti equilibrium around the glycosidic bond and therefore minimally perturbing the duplex DNA structure. A variety of tethers and tether lengths were considered to attach Gd3+-DOTA to the C5 position of uracil. However, as a proof of principle we reasoned that 5-hexyn-1-ol should be long enough to allow for on-resin post-synthetic click chemistry with alkyne Gd3+-DOTA. Shorter length linkers such as 3-butyn-1-ol could have placed the alkyl-azide in a sterically hindered environment thus inhibiting the click chemistry reaction.39 Conversely, a longer tether might result in a larger distance distribution in DEER-EPR due to residual motion of the Gd3+-DOTA. The synthesis of phosphoramidite 2 begins with 5′-DMT-5-iodo-2′-deoxyuridine (DMT-5-I-dU)40 and is accomplished in three steps (Scheme 1). Sonogashira cross-coupling41 between DMT-5-I-dU and 5-hexyn-1-ol gave 1 in 72%. Conversion of the hydroxyl group to the iodo group proceeded without difficulty using (PhO)3PCH3I in DMF, followed by phosphitylation using standard conditions to give the phosphoramidite building block 2 in 52% over two steps.
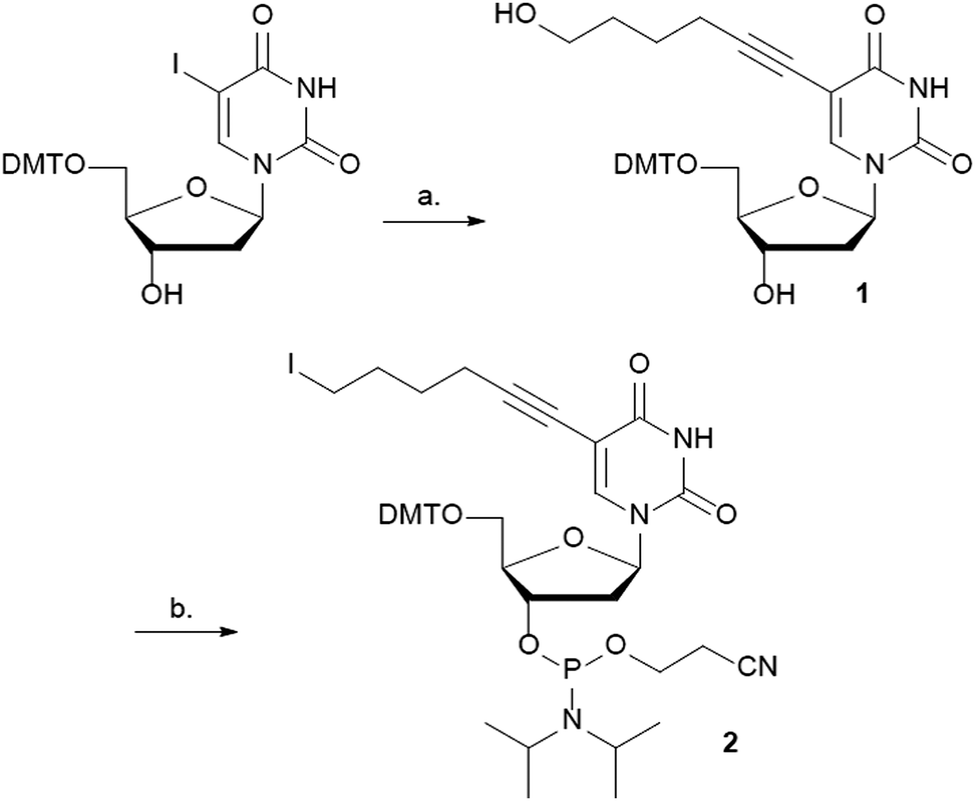 | ||
| Scheme 1 Synthesis of phosphoramidite 2. Reagents and conditions: (a) Pd(PPh3)2Cl2, CuI, NEt3, 5-hexyn-1-ol, THF, rt, 16 h, 72%; (b) (i) (PhO)3PCH3I, DIPEA, DMF, rt, 6 h, (ii) Cl-(i-Pr)2NPOCH2CH2CN, DIPEA, CH2Cl2, rt, 2 h, 52% (2 steps). | ||
Kool and co-workers have described that an iodothymidine phosphoramidite is stable towards standard automated DNA synthesis conditions.42 Furthermore, the iodo group may be easily displaced by various nucleophiles such as sodium azide.43,44 Based on these results, we reasoned that the iodo-containing phosphoramidite 2 would be compatible with DNA synthesis conditions and provide access to azide containing DNA.
Two modified oligodeoxynucleotides were synthesized DNA 1 5′-GCGAGTGACTGGTATGAXGATGCT-3′ and DNA 2 5′-AGCATCATCATACCAGXCACTCGC-3′ containing the modified building block X (Scheme 2). The iodo-containing resin-bound DNAs (DNA 1 or DNA 2) were converted to the corresponding azides using an excess of NaN3 at 50 °C. Alkyne-(Gd3+-DOTA) was prepared as described by M.G Finn and co-workers.37 On-resin click chemistry between the azide DNAs and alkyne-(Gd3+-DOTA) gave the desired Gd3+-DOTA containing DNAs, for details see the ESI.† The modified DNAs were deprotected under standard ammonia conditions followed by RP-HPLC purification, and characterization by ESI-MS. The purity was controlled via PAGE analysis and the labelling efficiency was calculated to be 80% via cw EPR.
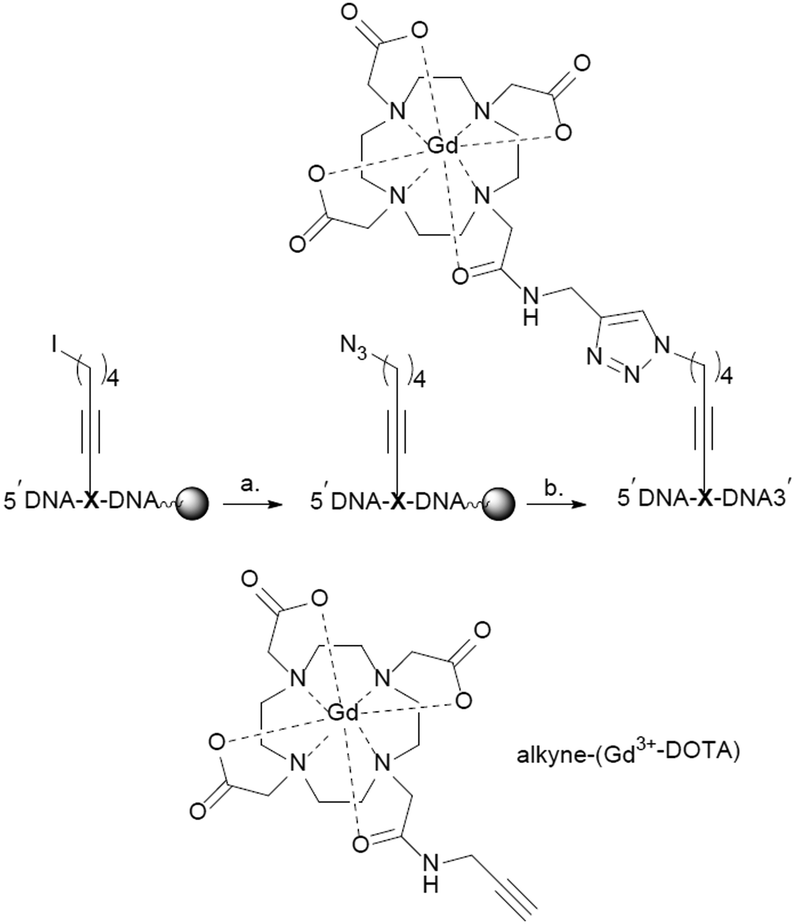 | ||
| Scheme 2 On-resin synthesis of internally labelled DNA. (a) NaN3, DMF, 55 °C. 24 h; (b) (i) TBTA, sodium ascorbate, CuSO4, alkyne-(Gd3+-DOTA), DMF/H2O, 50 °C, (ii) NH4OH, 50 °C, 12 h. X represents the modified nucleotide containing building block 2. | ||
Duplex DNA (DNA 1/DNA 2) resulted in a decrease in thermal stability (ΔTm = −10 °C, Tm = 54 °C) and a deviation from a two-state melting profile when compared to the duplex control sequence (Tm = 64 °C), Fig. 1a. Next we performed CD spectroscopy in order to gain insight into the structure of the DNA duplex. A typical B-type conformation with almost equal positive (275 nm) and negative (245 nm) ellipticity zeroing at 260 nm was observed (Fig. 1b). Altogether attaching the Gd-DOTA to DNA has a destabilizing effect when compared to the unmodified DNA as observed by a decrease in thermal stability. However CD spectroscopy indicates that the modified duplex exists as a B-DNA duplex.
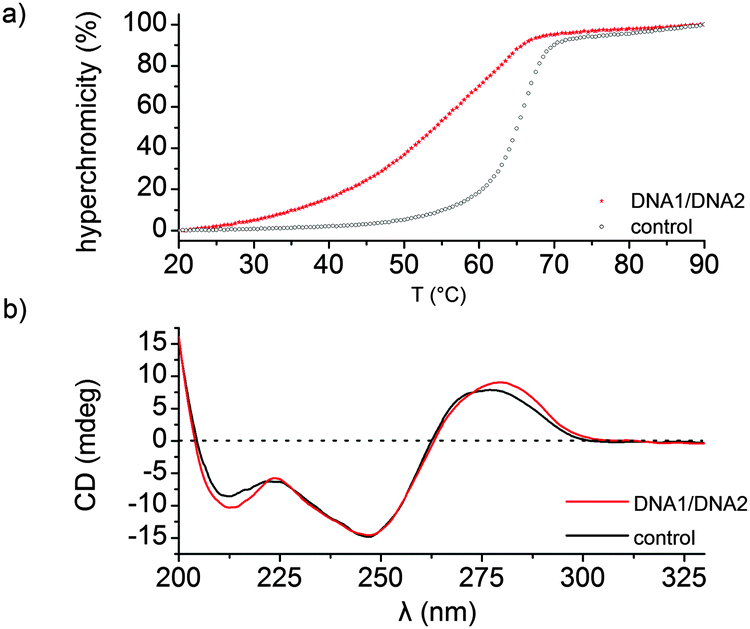 | ||
| Fig. 1 Thermal denaturation and CD spectra. (a) UV-thermal denaturation curve of the modified duplex DNA (DNA 1/DNA 2, Tm = 54 °C) and the control sequence (Tm = 64 °C). (b) CD spectra of duplex DNA 1/DNA 2 and the control sequence. | ||
Q-Band (34 GHz) EPR-experiments of the internally labelled duplex DNA were carried out in deuterated Tris-HCl buffer and inside Xenopus laevis oocytes (for in-cell samples see Fig. 2a). The oocytes for these samples were shock-frozen after 15 minutes of incubation time. Although Gd3+-complexes are stable in the cellular milieu for hours,9,10 we noticed a cytotoxic effect of the Gd3+ labelled duplex DNA for the oocytes. We observed the disruption of the vitelline membrane of the oocytes after 1 hour of incubation after microinjection. Even after 15 min of incubation signs of apoptosis were observable as loss of pigment in the animal pole. However, the vitelline membrane was still intact and no leakage was observed (see the ESI† for details). The central transition-normalized echo-detected field sweeps of the in vitro (black) and intracellular (orange) samples are shown in Fig. 3b. The spectral shape of both experiments are similar, whereas the intracellular measurement features a further superimposed sextet signal allocated to endogenous Mn2+ species.9,10
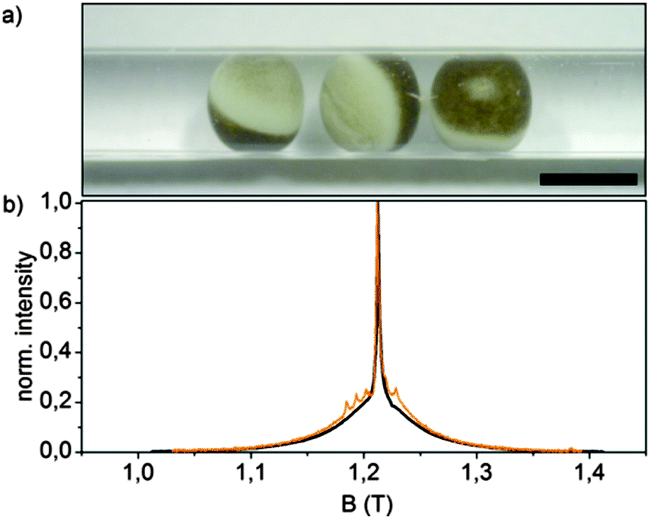 | ||
| Fig. 2 (a) Micrograph of a DEER-sample of 3 oocytes of Xenopus laevis in a Q-band test tube after 15 min incubation at 18 °C with 50 nL of 3.7 mM duplex DNA microinjected in each oocyte, scale bar = 1 mm; (b) normalized Q-band two-pulse echo-detected field sweeps of duplex DNA in buffer (black) and inside Xenopus laevis oocytes after 15 min incubation (orange), measured after shock-freezing at 10 K. | ||
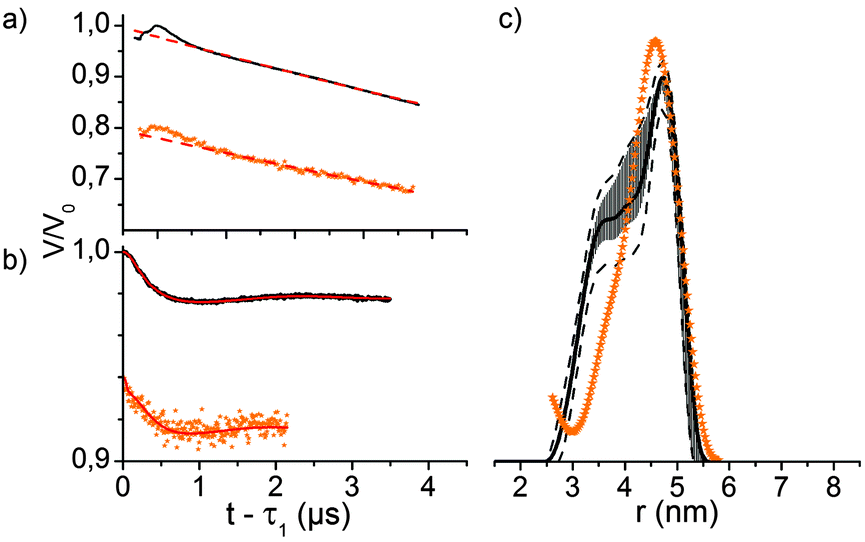 | ||
| Fig. 3 (a) Four-pulse DEER traces of duplex DNA in buffer (black) and inside Xenopus laevis oocytes after 15 min incubation (orange) recorded at the Q-band (T = 10 K) and corresponding background fits (dashed red lines), assuming a homogeneous 3D distribution of molecules; (b) background-corrected DEER form factor of the corresponding experiments with Tikhonov regularization fit (red); (c) distance distribution obtained from Tikhonov regularization of the corresponding measurements; observer pulses 14/14/28 ns, pump pulses 16 ns (buffer), 18 ns (in-cell), Δν = 125 MHz, accumulation time 17 h (buffer) or 20 h (in-cell); (c) distance distributions obtained via Tikhonov regularization of the corresponding DEER traces of dsDNA in buffer (black) and inside Xenopus laevis oocytes (orange). The error bars are the full variation of the probability of the given distance distribution (black) obtained during the statistical analysis, whereas the dotted lines are upper/lower error estimations corresponding to the mean value +/− two times the standard deviation of the different Tikhonov fits. | ||
Q-band-DEER-measurements of the internally labelled duplex DNA (100 μM) were carried out in deuterated Tris-HCl buffer (pH = 7.4) at T = 10 K. The label efficiency was calculated to be 80% by determination of spin concentration via EPR (echo-detected field sweep shown in Fig. 2b). Fig. 3a shows the DEER-curve (black) with 3-dimensional background function (red) and Fig. 3b shows the background corrected form factor (black) with model-free Tikhonov-regularization fit (red), respectively. The modulation depth λ = 2% is expected for DOTA-based labels under the applied conditions.20 The received distance distribution of the in vitro experiment is shown in Fig. 3c (black), featuring a mean distance of 4.8 nm. However, validation of these data via 160 Tikhonov-regularizations (shown as error bars as described in the Experimental section) reveals that the shoulder at 3.8 nm should not be over-interpreted. In spite of the rather long tether of the spin label, we find a distance distribution featuring a half width at half maximum of 0.83 nm, which is slightly broader as reported by Song et al. for a terminally Gd(III) labelled DNA.23
For EPR distance measurements of the duplex DNA inside Xenopus laevis 50 nL stock solution (3.7 mM DNA in Tris-HCl buffer) were microinjected into each cell. The average spin concentration in the sample determined by the echo-detected field sweep is 180 μM with a local concentration of 350 μM as calculated from the dipolar evolution curve. The corresponding DEER-curve and form factor are shown in Fig. 3a and b in orange. The modulation depth of the form factor is comparable with the modulation depth of the in vitro measurement, but the signal-to-noise is worse since the transverse relaxation time of T2 = 1.4 ms is significantly shorter than the transverse relaxation time of the in vitro measurement with T2 = 4.0 ms (see Fig. S2, ESI†). Furthermore, the signal-to-noise ratio of the DEER form factor is reduced by the endogenous, spatially 3-dimensionally distributed Mn2+ species which contributes solely to the measurement background, effectively reducing the signal-to-noise ratio.9 The distance distribution in cellula obtained by Tikhonov-regularization is shown in Fig. 3c. Due to the short dipolar evolution time of the measurement solely the mean distance of the distance distribution is reliable and the discussion of shape and width would involve the over-interpretation of the available experimental data. Still, the obtained mean distance fits quite well with the mean distance of the measurement in buffer, displaying the stability of the doubly internally labelled duplex DNA and of the label itself.
In conclusion we have introduced a straightforward method for labelling oligonucleotides internally with Gd3+-labels. The formerly utilized 5′-end could in principle be utilized for additional modifications. More importantly, internal labels give more flexibility in terms of the investigated structures. For example, individual domains or parts within the context of larger nucleic acid sequences can be characterized by internal labelling strategies. Although the stability of the investigated duplex DNA decreased, importantly the observed B-DNA structure was not disturbed. Moreover, the internal Gd labels allowed measurement of intermolecular distances of the labelled DNA duplex. An increasing field of interest is the application of EPR spectroscopy in cellular environments. For this purpose, modified biomolecules are introduced into cells and EPR measurements are carried out. Here, we demonstrate the utilization of internal Gd labels for acquiring distance measurements in Xenopus oocytes. Compared to nitroxide labels utilized so far, Gd labels have the advantage of increased intracellular stability. However, we observed pronounced cytotoxicity of the Gd-labelled DNAs that might prohibit investigations over extended times.
Notes and references
- G. Jeschke, Annu. Rev. Phys. Chem., 2012, 63, 419–446 CrossRef CAS PubMed.
- Y. Shin, C. Levinthal, F. Levinthal and W. Hubbell, Science, 1993, 259, 960–963 CrossRef CAS.
- D. T. Yu, A. D. Milov and A. G. Maryasov, Russ. Chem. Rev., 2008, 77, 487 Search PubMed.
- M. Azarkh, V. Singh, O. Okle, I. T. Seemann, D. R. Dietrich, J. S. Hartig and M. Drescher, Nat. Protoc., 2013, 8, 131–147 CrossRef CAS PubMed.
- M. Azarkh, O. Okle, V. Singh, I. T. Seemann, J. S. Hartig, D. R. Dietrich and M. Drescher, ChemBioChem, 2011, 12, 1992–1995 CrossRef CAS PubMed.
- M. Azarkh, O. Okle, P. Eyring, D. R. Dietrich and M. Drescher, J. Magn. Reson., 2011, 212, 450–454 CrossRef CAS PubMed.
- I. Krstić, R. Hänsel, O. Romainczyk, J. W. Engels, V. Dötsch and T. F. Prisner, Angew. Chem., Int. Ed., 2011, 50, 5070–5074 CrossRef PubMed.
- R. Igarashi, T. Sakai, H. Hara, T. Tenno, T. Tanaka, H. Tochio and M. Shirakawa, J. Am. Chem. Soc., 2010, 132, 8228–8229 CrossRef CAS PubMed.
- M. Qi, A. Groß, G. Jeschke, A. Godt and M. Drescher, J. Am. Chem. Soc., 2014, 136, 15366–15378 CrossRef CAS PubMed.
- A. Martorana, G. Bellapadrona, A. Feintuch, E. Di Gregorio, S. Aime and D. Goldfarb, J. Am. Chem. Soc., 2014, 136, 13458–13465 CrossRef CAS PubMed.
- M. Pannier, S. Veit, A. Godt, G. Jeschke and H. W. Spiess, J. Magn. Reson., 2000, 142, 331–340 CrossRef CAS PubMed.
- A. D. Milov, A. G. Maryasov and Y. D. Tsvetkov, Appl. Magn. Reson., 1998, 15, 107–143 CrossRef CAS.
- J. E. Banham, C. M. Baker, S. Ceola, I. J. Day, G. H. Grant, E. J. J. Groenen, C. T. Rodgers, G. Jeschke and C. R. Timmel, J. Magn. Reson., 2008, 191, 202–218 CrossRef CAS PubMed.
- V. Singh, M. Azarkh, T. E. Exner, J. S. Hartig and M. Drescher, Angew. Chem., Int. Ed., 2009, 48, 9728–9730 CrossRef CAS PubMed.
- I. T. Holder, M. Drescher and J. S. Hartig, Bioorg. Med. Chem., 2013, 21, 6156–6161 CrossRef CAS PubMed.
- C. Rehm, I. T. Holder, A. Groß, F. Wojciechowski, M. Urban, M. Sinn, M. Drescher and J. S. Hartig, Chem. Sci., 2014, 5, 2809–2818 RSC.
- P. M. Gannett, E. Darian, J. Powell, E. M. Johnson, C. Mundoma, N. L. Greenbaum, C. M. Ramsey, N. S. Dalal and D. E. Budil, Nucleic Acids Res., 2002, 30, 5328–5337 CrossRef CAS PubMed.
- U. Jakobsen, S. A. Shelke, S. Vogel and S. T. Sigurdsson, J. Am. Chem. Soc., 2010, 132, 10424–10428 CrossRef CAS PubMed.
- D. Goldfarb, Phys. Chem. Chem. Phys., 2014, 16, 9685–9699 RSC.
- A. M. Raitsimring, C. Gunanathan, A. Potapov, I. Efremenko, J. M. L. Martin, D. Milstein and D. Goldfarb, J. Am. Chem. Soc., 2007, 129, 14138–14139 CrossRef CAS PubMed.
- H. Yagi, D. Banerjee, B. Graham, T. Huber, D. Goldfarb and G. Otting, J. Am. Chem. Soc., 2011, 133, 10418–10421 CrossRef CAS PubMed.
- P. Lueders, H. Jäger, M. A. Hemminga, G. Jeschke and M. Yulikov, J. Phys. Chem. B, 2013, 117, 2061–2068 CrossRef CAS PubMed.
- Y. Song, T. J. Meade, A. V. Astashkin, E. L. Klein, J. H. Enemark and A. Raitsimring, J. Magn. Reson., 2011, 210, 59–68 CrossRef CAS PubMed.
- M. Bottrill, L. Kwok and N. J. Long, Chem. Soc. Rev., 2006, 35, 557–571 RSC.
- G. J. Stasiuk and N. J. Long, Chem. Commun., 2013, 49, 2732–2746 RSC.
- T. P. Gazzi, L. A. Basso, D. S. Santos and P. Machado, RSC Adv., 2014, 4, 9880–9884 RSC.
- L. Lattuada, A. Barge, G. Cravotto, G. B. Giovenzana and L. Tei, Chem. Soc. Rev., 2011, 40, 3019–3049 RSC.
- N. Viola-Villegas and R. P. Doyle, Coord. Chem. Rev., 2009, 253, 1906–1925 CrossRef CAS PubMed.
- P. A. Bonnet, A. Michel, J. P. Fernandez, C. Cyteval, A. Rifai, M. Boucard, J. P. Chapat and J. L. Lamarque, Magn. Reson. Imaging, 1990, 8, 71–77 CrossRef CAS.
- Y. Song, T. J. Meade, A. V. Astashkin, E. L. Klein, J. H. Enemark and A. Raitsimring, J. Magn. Reson., 2011, 210, 59–68 CrossRef CAS PubMed.
- J. Hovinen, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 1459–1462 CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388–1405 RSC.
- M. Gerowska, L. Hall, J. Richardson, M. Shelbourne and T. Brown, Tetrahedron, 2012, 68, 857–864 CrossRef CAS PubMed.
- T. Wada, A. Mochizuki, S. Higashiya, H. Tsuruoka, S. Kawahara, M. Ishikawa and M. Sekine, Tetrahedron Lett., 2001, 42, 9215–9219 CrossRef CAS.
- J. D. E. Prasuhn, R. M. Yeh, A. Obenaus, M. Manchester and M. G. Finn, Chem. Commun., 2007, 1269–1271 RSC.
- R. F. H. Viguier and A. N. Hulme, J. Am. Chem. Soc., 2006, 128, 11370–11371 CrossRef CAS PubMed.
- J. Gierlich, G. A. Burley, P. M. E. Gramlich, D. M. Hammond and T. Carell, Org. Lett., 2006, 8, 3639–3642 CrossRef CAS PubMed.
- R. H. E. Hudson and A. Ghorbani-Choghamarani, Synlett, 2007, 0870–0873 CrossRef CAS.
- K. Sonogashira, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS.
- Y. Xu and E. T. Kool, Tetrahedron Lett., 1997, 38, 5595–5598 CrossRef CAS.
- G. P. Miller and E. T. Kool, J. Org. Chem., 2004, 69, 2404–2410 CrossRef CAS PubMed.
- G. P. Miller and E. T. Kool, Org. Lett., 2002, 4, 3599–3601 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional experiments. See DOI: 10.1039/c5cc04234h |
| This journal is © The Royal Society of Chemistry 2015 |