
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a PtSn bimetallic catalyst for direct fuel cells using bio-butanol fuel†
V. K.
Puthiyapura
a,
D. J. L.
Brett
b,
A. E.
Russell
c,
W. F.
Lin
*a and
C.
Hardacre
*a
aCentre for the Theory and Application of catalysis (CenTACat), School of Chemistry and Chemical Engineering, Queens University of Belfast, Belfast-BT95AG, UK. E-mail: w.lin@qub.ac.uk; c.hardacre@qub.ac.uk
bElectrochemical Innovation Lab, Department of Chemical Engineering, University College London, Torrington Place, London WC1E 7JE, UK
cDepartment of Chemistry, University of Southampton, High field, Southampton-SO17 1BJ, UK
First published on 20th July 2015
Abstract
Pt and PtSn catalysts were studied for n-butanol electro-oxidation at various temperatures. PtSn showed a higher activity towards butanol electro-oxidation compared to Pt in acidic media. The onset potential for n-butanol oxidation on PtSn is ∼520 mV lower than that found on Pt, and significantly lower activation energy was found for PtSn compared with that for Pt.
Low molecular weight alcohols, such as methanol and ethanol, have been proposed as promising alternative fuels to H2 in low-temperature fuel cells due to their liquid nature, high energy density,1 low toxicity, availability and ease of handling.1a,2 Methanol and ethanol are widely studied as fuels for direct alcohol fuel cells (DAFCs). Methanol is toxic, whereas ethanol is non-toxic and can be produced by fermentation of sugar. However, there is an on-going public debate on the ethics of utilizing food stocks for fuel production instead of nutrition.3 An alternate renewable source, which does not compete with food, is required for sustainable long-term energy production. Thus, second generation bio-fuels are proposed to be produced from non-food based biomass feedstock such as lignocellulose biomass (LCB).3,4n-Butanol (n-BtOH) is considered as a 2nd generation bio-fuel, with better infrastructure compatibility than ethanol; it has higher energy density, lower water adsorption, and better blending ability with gasoline than ethanol and is considered as a potential substitute for gasoline in automobile fuel.3,4 Also, bio-butanol is non-poisonous, non-corrosive, bio-degradable and does not lead to soil and water pollution.5 Multiple companies are working on the development of commercial scale bio-butanol production. For example, GreenBiologics UK,6 and Cobalt Technologies USA7 are involved in bio-butanol production. Butamax,8 a joint venture between BP and DuPont, has developed technology to produce bio-butanol from food stock biomass8 and in the second phase, is planning to produce butanol from LCB.8a Syntec Biofuels, Canada,9 uses a thermo-chemical method to produce bio-butanol from waste biomass (municipal solid waste, agricultural and forestry wastes).
Although there are a significant number of studies examining small-chain alcohols, such as methanol and ethanol, as potential fuels for fuel cells, there are fewer studies on longer-chain alcohols such as butanol. On complete electro-oxidation, butanol can provide 24e− per molecule compared to 12e− and 6e− for ethanol and methanol, respectively (eqn (1) for the general electro-oxidation of mono alcohols). However, increasing the number of carbon chain will increase the number of possible intermediates and complicate the reaction pathway.10
| CnH2n+1OH + (2n − 1)H2O → nCO2 + 6nH+ + 6ne− | (1) |
Butanol also has an advantage over shorter chain length alcohols in that the fuel cross-over through the electrolyte membrane, which is one of the major issues in DAFCs, will be reduced. Takky et al.11 studied butanol electro-oxidation on different electrodes (Pt, Au, Rh, Pd) in alkaline media. They have observed that n-butanol and iso-butanol show similar activity, whereas 2-butanol was less reactive. In contrast, tert-butanol was not oxidizable at all at room temperature. Only n-butanol and iso-butanol are produced from biomass feed stocks. Of the two, n-butanol appears to be the most promising as a potential fuel for DAFCs. In the work reported in this communication, the electro-oxidation of n-butanol is explored at both Pt and bimetallic PtSn electrocatalysts, building upon the previously published studies, which only described the monometallic catalyst.10a
The electrochemical measurements were run using a home built water-jacket three-electrode cell at controlled temperatures using a Bio-Logic EC Lab SP-200 potentiostat connected to a PC with EC-Lab software. A Ag/AgCl in 3 M NaCl (BASi, USA) (0.210 V vs. SHE at 25 °C) reference electrode was used, and a Pt mesh (Goodfellow, UK) attached to a Pt wire (Mateck, Germany) was used as counter electrode. All potentials are reported with respect to Ag/AgCl – 3 M NaCl unless otherwise specified. Pt was electro-deposited on a glassy carbon (GC) substrate (area = 0.38 cm2) from 5 mM H2PtCl6 + 0.1 M HCl aqueous solution and Sn was electro-deposited on the Pt from 1mM SnCl2 + 0.5 M H2SO4 solution at a deposition potential of −0.210 V. The Sn coverage can be controlled by Sn deposition time; for example, the 25% Sn coverage can be achieved in 20 seconds. The electrodeposited Pt has ‘cauliflower-like’ spherical morphology and no significant morphology change occurred upon the Sn deposition on the Pt from the SEM analysis (ESI,† Fig. S1 and S2). The cyclic voltammogram (CV) of Pt showed characteristic features of polycrystalline Pt in acidic solution (ESI,† Fig. S3) and the Pt active area (Ar) was calculated using eqn (2).
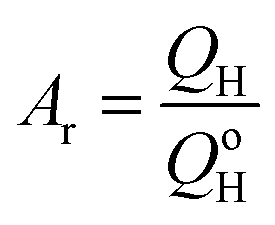 | (2) |
 | ||
| Fig. 1 The cyclic voltammogram of Pt/GC in 0.1M BtOH + 0.1 M H2SO4 solution. Scan rate 20 mV s−1. | ||
It is noted that the onset of n-butanol oxidation, peak a1, intersects with that of the water oxidation on Pt (ESI,† Fig. S3) indicating that the formation of the surface OHads may facilitate n-butanol oxidation on Pt. Increasing the potential promotes the formation of surface OHads which in turn enhances the n-butanol oxidation (peak a2). However, at potentials >1.0 V the Pt surface is covered by the oxide species leading to a decrease in n-butanol oxidation current.12 The reduction of the oxide species during the NGPS allows fresh adsorption and oxidation of butanol giving peak a3.
At higher temperatures, a significant improvement in n-butanol oxidation was observed. An increase in current with a negative shift in the onset potential and a positive shift in the re-oxidation peak was observed with the increase in temperature (Fig. 2). The activation energy (Ea) for the pure Pt in the n-butanol oxidation calculated from the Arrhenius plot (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j vs. 1/T) was ∼25 kJ mol−1 and ∼15 kJ mol−1 at 0.70 V and 1.00 V, respectively (Fig. 3a). A higher activation energy for the first anodic peak compared to the second anodic peak was reported in literature for alcohol oxidation reaction on Pt in acidic media.12,13
j vs. 1/T) was ∼25 kJ mol−1 and ∼15 kJ mol−1 at 0.70 V and 1.00 V, respectively (Fig. 3a). A higher activation energy for the first anodic peak compared to the second anodic peak was reported in literature for alcohol oxidation reaction on Pt in acidic media.12,13
 | ||
| Fig. 2 The cyclic voltammograms showing the effect of temperature on the butanol oxidation on Pt/GC electrode in 0.1 M n-BtOH + 0.1 M H2SO4 solution. Scan rate 20 mV s−1. | ||
 | ||
| Fig. 3 Arrhenius plots for (a) Pt in 0.1 M n-BtOH + 0.1 M H2SO4 at 0.70 V and 1.00 V and (b) PtSn/GC in 0.1 M n-BtOH + 0.1 M H2SO4 at 0.25 V (θSn = ∼17%). | ||
It is well-documented that addition of oxophilic metals such as Ru and Sn to Pt can promote surface oxidant OHads formation at lower potential. This is expected to enhance the alcohol oxidation through the bi-functional mechanism.14 PtRu is known to be the best catalyst for the methanol oxidation reaction, whilst PtSn has been reported to be the most active bimetallic catalyst for ethanol.10b,15,16
A PtSn electrode was prepared by electrochemical deposition of Sn on Pt/GC. The CVs of Pt and PtSn electrodes in 0.1 M H2SO4 supporting electrolyte and n-butanol containing solutions are compared in Fig. 4. The upper potential in CV analysis was limited to 0.75 V in the case of the PtSn electrode in order to avoid any electro-dissolution of Sn at higher potentials.17 The hydrogen adsorption/desorption region was suppressed on Sn addition due to the blockage of the Pt sites by Sn for hydrogen adsorption. The Sn coverage (θSn) on Pt was calculated from the change in hydrogen desorption charge before and after the Sn deposition on Pt/GC according to eqn (3), where QSnH is the hydrogen desorption charge for the PtSn electrode and QH is the hydrogen desorption charge for Pt.10b,18 The θSn calculated from the CV for the PtSn was ∼25%.
 | ||
| Fig. 4 (a) Cyclic voltammograms of Pt and PtSn electrode in 0.1 M H2SO4 and (b) PGPS voltammograms of Pt and PtSn in 0.1 M n-BtOH + 0.1 M H2SO4. Scan rate 50 mV s−1. Sn coverage (θSn) ∼25%. | ||
For the PtSn in H2SO4 solution (see Fig. 4a), surface oxidation appears with an onset potential of ∼0.10 V and peak potential at around 0.55 V indicating the oxidation of the adsorbed Sn.18 The hydrogen adsorption/desorption features are preserved for the PtSn electrodes but with reduced area.
Fig. 4b compares the PGPS voltammograms of the Pt and PtSn electrodes in 0.1 M n-BtOH + 0.1 M H2SO4. The Pt shows the characteristic features in acid19 with a n-BtOH oxidation onset potential of ∼0.55 V. A similar onset potential was reported in literature for n-butanol oxidation on Pt in acidic media.1a However, for the PtSn electrode, a pre-peak at ∼0.27 V (onset potential at ∼0.03 V) was observed before the a1 peak on Pt. This peak was only observed in the n-butanol containing solution, indicating that the peak originated from butanol oxidation. This was further confirmed by adding butanol gradually to the sulphuric acid supporting electrolyte while running the CV and the pre-peak appears only when butanol is added to the acidic solution. In fact, Sn coverage of 10–30% was found to be optimum (ESI,† Fig. S4). At higher Sn coverage (θSn > 40%), the butanol oxidation peak a1 was found to be suppressed significantly (ESI,† Fig. S5). Since this pre-peak appears in a potential region far below PtOH/PtOx formation, the active oxygen species for the n-butanol oxidation must originate from the Sn and is probably due to the low onset potential for OH formation on Sn observed previously.10b,14,20
 | (3) |
In order to have an understanding of the oxidation pre-peak behaviour with other alcohol molecules, PtSn for ethanol (EtOH) and iso-butanol (iso-BtOH) (ESI,† Fig. S6) were compared with that of n-butanol. The pre-peak was also present in both iso-BtOH and EtOH solution. It is interesting to note that even though the onset and half wave potentials of ethanol oxidation22 (∼0.40 V and ∼0.56 V, respectively) and n-butanol oxidation (∼0.55 V and ∼0.64 V) were different on pure Pt, the onset and half wave potentials for all alcohols were the same for PtSn (∼0.02 V and ∼0.16–0.20 V respectively) (Table 1). This result is significant as it indicates that on PtSn the oxidation is initiated and determined by the water oxidation on Sn, rather than on the nature of the molecule. González et al.23 observed that alcohols with H atoms on the β carbon can be oxidized on PtSn electrodes at lower potential than on monometallic Pt electrode. They proposed that H abstraction from α and β carbon could lead to the formation of a stabilizing enol structure, which could convert to aldehyde. This conversion would be facilitated by the OH/O-rich PtSn surface as the attack on enol by adsorbed H2O could easily form a hydrated aldehyde.23
| Catalyst | Onset potential and (E1/2)/V | ||
|---|---|---|---|
| Butanol | iso-Butanol | Ethanol | |
| Pt | ∼0.55 | ∼0.55 | ∼0.40 |
| (∼0.64) | (∼0.62) | (∼0.56) | |
| PtSn | ∼0.03 | ∼0.03 | ∼0.03 |
| (∼0.16) | (∼0.16) | (∼0.20) | |
In conclusion, significantly higher activity of a PtSn bimetallic catalyst for n-butanol electro-oxidation in acidic media compared to pure Pt has been clearly demonstrated. On addition of Sn, a distinguishable oxidation pre-peak was observed at a much lower onset potential for PtSn compared to those for Pt. This has been attributed to facile formation of OHads at lower potentials on Sn. These results suggest that PtSn could be an effective catalyst for n-butanol electro-oxidation in direct alcohol fuel cells.
The authors acknowledge the financial support from the EPSRC (EP/K014706/1).
Notes and references
- (a) C. Lamy, E. M. Belgsir and J. M. Léger, J. Appl. Electrochem., 2001, 31, 799 CrossRef CAS; (b) A. Brouzgou, F. Tzorbatzoglou and P. Tsiakaras, Energetics (IYCE), Proceedings of the 2011 3rd International Youth Conference, 2011, pp. 1–6 Search PubMed.
- Z. X. Liang and T. S. Zhao, Catalysts for Alcohol-fuelled Direct Oxidation Fuel Cells, Royal Society of Chemistry, 2012 Search PubMed.
- P. Dürre, Biotechnol. J., 2007, 2, 1525 CrossRef PubMed.
- L. Tao, X. He, E. C. D. Tan, M. Zhang and A. Aden, Biofuels, Bioprod. Biorefin., 2014, 8, 342 CrossRef CAS PubMed.
- P. Patakova, D. Maxa, M. Rychtera, M. Linhova, P. Fribert, Z. Muzikova, J. Lipovsky, L. Paulova, M. Pospisil and G. S. a. K. Melzoch, in Biofuel's Engineering Process Technology, ed. M. A. D. S. Bernardes, InTech, 2011 Search PubMed.
- Green Biologics Ltd (GBL), http://www.greenbiologics.com/index.php, accessed February 2015.
- Cobalt Technologies, http://www.cobalttech.com/technology.html, accessed February 2015, 2015.
- (a) L. Butamax™ Advanced Biofuels, BP and DuPont, 2010; (b) Butamax™, http://www.butamax.com/, accessed February 2015.
- Syntec Biofuel Inc, http://www.syntecbiofuel.com/index.php, accessed February 2015, 2015.
- (a) N. H. Li and S. G. Sun, J. Electroanal. Chem., 1997, 436, 65 CrossRef CAS; (b) J. M. Jin, T. Sheng, X. Lin, R. Kavanagh, P. Hamer, P. Hu, C. Hardacre, A. Martinez-Bonastre, J. Sharman, D. Thompsett and W. F. Lin, Phys. Chem. Chem. Phys., 2014, 16, 9432 RSC; (c) Z. G. Shao, F. Y. Zhu, W. F. Lin, P. A. Christensen, H. M. Zhang and B. L. Yi, J. Electrochem. Soc., 2006, 153, A1575 CrossRef CAS PubMed.
- (a) D. Takky, B. Beden, J. M. Leger and C. Lamy, J. Electroanal. Chem. Interfacial Electrochem., 1988, 256, 127 CrossRef CAS; (b) D. Takky, B. Beden, J. M. Leger and C. Lamy, J. Electroanal. Chem. Interfacial Electrochem., 1983, 145, 461 CrossRef CAS; (c) D. Takky, B. Beden, J.-M. Leger and C. Lamy, J. Electroanal. Chem. Interfacial Electrochem., 1985, 193, 159 CrossRef CAS.
- C.-G. Lee, M. Umeda and I. Uchida, J. Power Sources, 2006, 160, 78 CrossRef CAS PubMed.
- S. N. Raicheva, M. V. Christov and E. I. Sokolova, Electrochim. Acta, 1981, 26, 1669 CrossRef CAS.
- S. S. Gupta, S. Singh and J. Datta, Mater. Chem. Phys., 2009, 116, 223 CrossRef PubMed.
- (a) J. Flórez-Montaño, G. García, J. L. Rodríguez, E. Pastor, P. Cappellari and G. A. Planes, J. Power Sources, 2015, 282, 34 CrossRef PubMed; (b) Z. D. Wei, L. L. Li, Y. H. Luo, C. Yan, C. X. Sun, G. Z. Yin and P. K. Shen, J. Phys. Chem. B, 2006, 110, 26055 CrossRef CAS PubMed.
- F. Vigier, C. Coutanceau, F. Hahn, E. M. Belgsir and C. Lamy, J. Electroanal. Chem., 2004, 563, 81 CrossRef CAS PubMed.
- H. Li, G. Sun, L. Cao, L. Jiang and Q. Xin, Electrochim. Acta, 2007, 52, 6622 CrossRef CAS PubMed.
- E. Lamy-Pitara, L. E. Ouazzani-Benhima, J. Barbier, M. Cahoreau and J. Caisso, J. Electroanal. Chem., 1994, 372, 233 CrossRef CAS.
- D. R. Lowde, J. O. Williams and B. D. McNicol, Appl. Surf. Sci., 1978, 1, 215 CrossRef CAS.
- S. Beyhan, C. Coutanceau, J.-M. Léger, T. W. Napporn and F. Kadırgan, Int. J. Hydrogen Energy, 2013, 38, 6830 CrossRef CAS PubMed.
- S. Sen Gupta, S. Singh and J. Datta, Mater. Chem. Phys., 2010, 120, 682 CrossRef CAS PubMed.
- H. Razmi, E. Habibi and H. Heidari, Electrochim. Acta, 2008, 53, 8178 CrossRef CAS PubMed.
- M. J. González, C. T. Hable and M. S. Wrighton, J. Phys. Chem. B, 1998, 102, 9881 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc04188k |
| This journal is © The Royal Society of Chemistry 2015 |