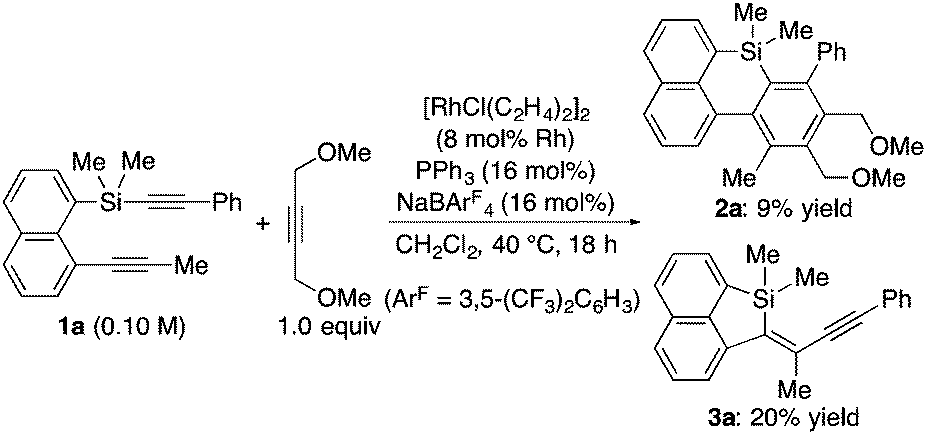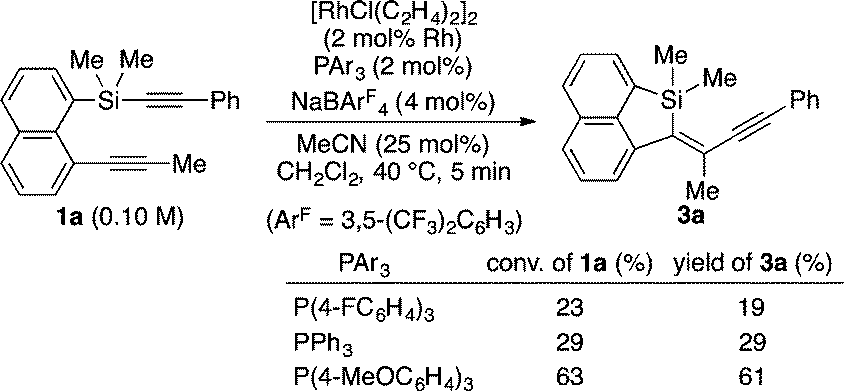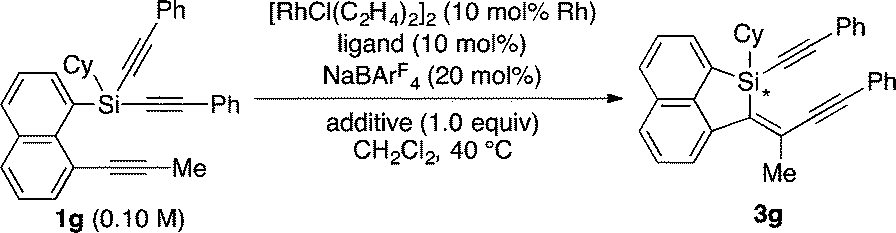DOI:
10.1039/C5CC04172D
(Communication)
Chem. Commun., 2015,
51, 11378-11381
Rhodium-catalyzed intramolecular alkynylsilylation of alkynes†
Received
20th May 2015
, Accepted 4th June 2015
First published on 4th June 2015
Abstract
Rhodium-catalyzed intramolecular alkynylsilylation of alkynes is described. The reaction proceeds through syn-insertion by a cationic rhodium/triarylphosphine catalyst, representing the first alkynylsilylation of alkynes via the cleavage of a C(sp)–Si bond by transition-metal catalysis. A highly enantioselective variant is also described for the creation of a silicon stereogenic center.
Stereoselective insertion of alkynes into carbon–silicon bonds represents a powerful and efficient approach for the synthesis of highly substituted alkenylsilanes. Most of the reported examples employ either strained organosilicon substrates1 or (Lewis) acid catalysts/additives2 to promote the reaction to achieve alkyl-,1a–f allyl-,1g–i,2b–e,3 alkenyl-,1g–j,2f–i,4 aryl-,1b,k,2h,i and propargyl/allenylsilylation2j of alkynes. More reactive acylsilanes5 and trimethylsilylcyanide6 are also known to undergo insertion of alkynes to give 2-acyl- and 2-cyanoalkenylsilanes, respectively. In contrast, virtually no progress has been made for alkyne insertion into alkynylsilanes. In fact, there has been only one report where they described a formal insertion reaction through conjugate addition of 2-silylynamides to acetylenedicarboxylates followed by silyl migration.7,8 In this communication, we describe the development of rhodium-catalyzed intramolecular alkynylsilylation of alkynes through the cleavage of a C(sp)–Si bond under mild conditions, including a highly enantioselective variant for the construction of a silicon stereogenic center.9
During the course of our study directed toward the development of synthetic methods for various silicon-bridged π-conjugated compounds,10 we attempted to synthesize benzonaphthosiline 2a from silicon-containing diyne 1a and 1,4-dimethoxy-2-butyne through a rhodium-catalyzed [2+2+2] cycloaddition reaction.11 As shown in eqn (1), under the conditions of using a cationic Rh/2PPh3 catalyst generated in situ, only 9% yield of the desired product 2a was obtained and the major product turned out to be an intramolecular alkynylsilylation product 3a in 20% yield. On the basis of this unexpected result, we decided to focus on the improvement of this alkynylsilylation reaction by rhodium catalysis. To our surprise, however, simple removal of 1,4-dimethoxy-2-butyne from the reaction in eqn (1) did not provide 3a at all (eqn (2)). We hypothesized that this seemingly inconsistent result might indicate that the coordination of 1,4-dimethoxy-2-butyne to rhodium had a beneficial effect on promoting the alkynylsilylation of 1a. We then tried to search for an innocent replacement and found that the use of MeCN as an additive gave product 3a in 51% yield, and a higher yield of 82% was achieved by changing the ratio of Rh/PPh3 from 1/2 to 1/1.12
| |  | (1) |
| |  | (2) |
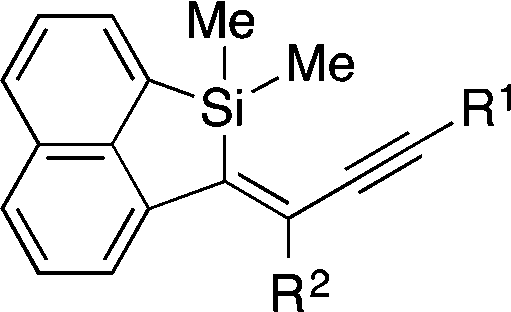
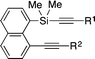
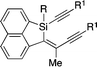
Under these conditions, several different alkynyl groups on the silicon atom of 1 are tolerated in the present intramolecular alkynylsilylation reaction to give the corresponding 3 in high yields, although an elevated temperature is necessary for the reaction of 1-propynyl substituted substrate 1d (Table 1, entries 1–4). The structure of 3b was confirmed by X-ray crystallographic analysis, establishing the syn-insertion of an alkyne into the alkynylsilane in the present catalysis.13 With respect to the alkynyl substituent at the 8-position of the naphthalene tether, in addition to alkyl groups such as 1a and 1e, aryl groups such as 1f can also be effectively employed by changing the ligand from PPh3 to P(4-MeOC6H4)3 (entries 1, 5 and 6). Substrates 1g–1i having an alkylbis(alkynyl)silyl group at the 1-position are also suitable for the present alkynylsilylation to give 3g–3i in 73–93% yield (entries 7–9). Furthermore, the reaction is applicable to substrates containing some other tethers as well. As shown in eqn (3) and (4), 1,2-bis(dimethyl(phenylethynyl)silyl)benzene (1j) and 1,8-bis(dimethyl(phenylethynyl)silyl)naphthalene (1k) similarly undergo intramolecular alkynylsilylation to give products 3j and 3k in high yields. The reaction also proceeds smoothly with substrate 4 having two alkynylsilane moieties through the two-fold alkynylsilylation process, giving a highly conjugated product 5 in 78% yield (eqn (5)).14
| |  | (3) |
| | 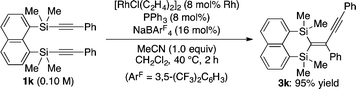 | (4) |
| |  | (5) |
Table 1 Rhodium-catalyzed alkynylsilylation: scopea
| Entry |
Substrate |
Product |
Yieldb (%) |
|
Conditions: [RhCl(C2H4)2]2 (8 mol% Rh), PPh3 (8 mol%), NaBAr4F (16 mol%), MeCN (1.0 equiv.), CH2Cl2 (0.10 M), 40 °C, 16 h.
Isolated yield (Z-isomer was obtained exclusively unless otherwise noted).
The reaction was conducted at 0.05 M substrate concentration.
Z/E = 98/2.
The reaction time was 1 h.
The reaction was conducted at 80 °C in toluene.
The reaction time was 2 h.
The reaction was conducted with P(4-MeOC6H4)3 instead of PPh3.
Z/E = 94/6.
|
|
|
|

|
|
| 1 |
1a (R1 = Ph, R2 = Me) |
3a
|
82 |
| 2c |
1b (R1 = 4-MeOC6H4, R2 = Me) |
3b
|
90d |
| 3e |
1c (R1 = 4-ClC6H4, R2 = Me) |
3c
|
93 |
| 4f |
1d (R1 = R2 = Me) |
3d
|
82 |
| 5g |
1e (R1 = Ph, R2 = n-Pr) |
3e
|
84 |
| 6c,h |
1f (R1 = R2 = Ph) |
3f
|
82i |
|
|

|
|
|
| 7g |
1g (R = Cy, R1 = Ph) |
3g
|
93 |
| 8c,e |
1h (R = Cy, R1 = Me) |
3h
|
82 |
| 9c,h |
1i (R = R1 = Me) |
3i
|
73 |
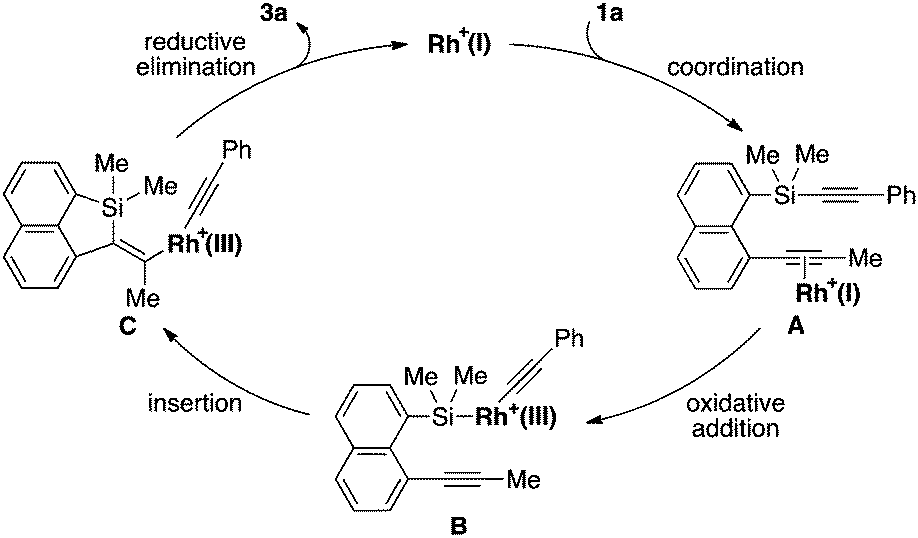
A proposed catalytic cycle for the reaction of 1a to 3a is illustrated in Scheme 1. Coordination of 1a to cationic rhodium(I) in the form of A facilitates the oxidative addition of a C(sp)–Si bond to give intermediate B. Successive intramolecular insertion of alkyne into a Si–Rh bond provides alkenyl(alkynyl)rhodium(III) C, reductive elimination of which leads to the formation of product 3a along with regeneration of the cationic rhodium(I) species. Although the role of MeCN is not yet completely understood, it probably stabilizes coordinatively unsaturated rhodium intermediates during the catalytic cycle. To gain some insights into the present catalysis, we conducted the following control experiments as shown in eqn (6) and (7). By changing the electronic properties of the triarylphosphine ligand in the reaction of 1a, we determined that the reaction proceeds faster by using more electron-rich phosphine ligands (eqn (6)). We also found that electron-deficient alkynylsilanes tend to react faster by changing the para-substituent of the arylethynyl group on the silicon of 1 (eqn (7)). Both these results are consistent with the assumption that the oxidative addition is the turnover-limiting step in the catalytic cycle, although further evidence is necessary to fully establish the catalytic cycle.
| | 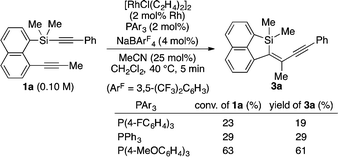 | (6) |
| |  | (7) |
 |
| | Scheme 1 Proposed catalytic cycle for the rhodium-catalyzed intramolecular alkynylsilylation of 1a. | |
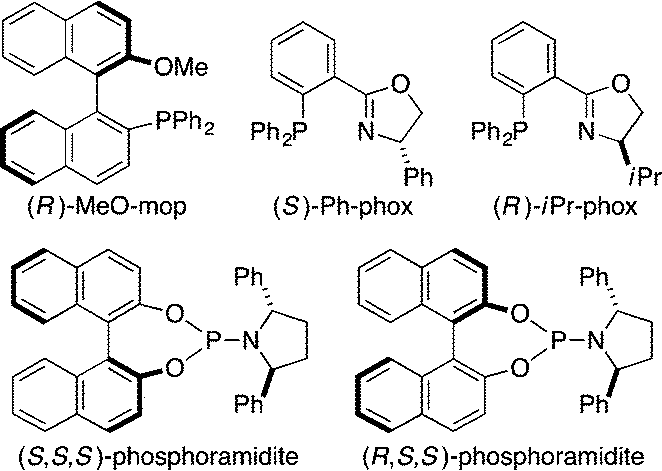
Finally, we have also begun to develop an asymmetric variant of this process.15 On the basis of the conditions for the nonasymmetric reactions, we conducted a reaction of prochiral 1g by employing (R)-MeO-mop,16 a chiral monophosphine ligand, in the presence of 1.0 equiv. of MeCN. Under these conditions, 77% yield of 3g was obtained, but no asymmetric induction was observed at the silicon stereocenter (Table 2, entry 1). In comparison, 83% ee was achieved with the same ligand in the absence of MeCN, but the yield of 3g became significantly lower (entry 2). To accommodate the nitrogen coordination to the structure of a chiral ligand, we examined (S)-Ph-phox,17 a P,N-bidentate ligand, and found that 3g was produced in 74% yield in the absence of MeCN with an appreciable ee of 69% (entry 3). Unfortunately, however, further improvement was unsuccessful by using other phosphinooxazoline ligands such as (R)-iPr-phox17 (entry 4). As a different structural motif for the chiral ligand, we also employed phosphoramidite ligands.18 For example, the use of (S,S,S)-phosphoramidite having a 2,5-diphenylpyrrolidine moiety19 gave 3g in 60% yield with 36% ee in the absence of MeCN (entry 5). We subsequently found that significantly higher enantioselectivity (92% ee) could be achieved by changing the ligand to its diastereomer ((R,S,S)-phosphoramidite)19a,20 with a moderate yield of 53% (entry 6). Both the yield and the ee were slightly improved further by lowering the initial substrate concentration from 0.10 M to 0.03 M to give 62% yield (59% isolated yield) of 3g with 94% ee (entry 7).
Table 2 Rhodium-catalyzed asymmetric alkynylsilylation
|

|
| Entry |
Ligand |
Additive |
Yielda (%) |
eeb (%) |
|
Determined by 1H NMR against an internal standard.
Determined by chiral HPLC on a Chiralcel OD-H column with hexane/2-propanol = 98/2.
The reaction was conducted at 0.03 M substrate concentration.
Isolated yield in parentheses.
|
| 1 |
(R)-MeO-mop |
MeCN |
77 |
0 |
| 2 |
(R)-MeO-mop |
None |
23 |
83 (−) |
| 3 |
(S)-Ph-phox |
None |
74 |
69 (+) |
| 4 |
(R)-iPr-phox |
None |
27 |
42 (−) |
| 5 |
(S,S,S)-Phosphoramidite |
None |
60 |
36 (+) |
| 6 |
(R,S,S)-Phosphoramidite |
None |
53 |
92 (+) |
| 7c |
(R,S,S)-Phosphoramidite |
None |
62 (59)d |
94 (+) |
In summary, we have developed rhodium-catalyzed intramolecular alkynylsilylation of alkynes under mild conditions. The reaction proceeds through syn-insertion in the presence of a cationic rhodium/triarylphosphine catalyst with MeCN as an additive. Although applicable substrates are currently still limited, this represents the first alkynylsilylation of alkynes via the cleavage of a C(sp)–Si bond by transition-metal catalysis. We have also described our preliminary investigation of its asymmetric variant, creating a silicon stereogenic center with high enantioselectivity. Future studies will be directed toward the development of a more general catalyst system to expand the scope of alkynylsilylation of alkynes and related reactions.
Support has been provided in part by Challenging Exploratory Research, the Ministry of Education, Culture, Sports, Science and Technology, Japan. We thank Prof. Takuzo Aida and Dr Yoshimitsu Itoh at The University of Tokyo for the measurement of fluorescence spectra.
Notes and references
-
(a) H. Sakurai and T. Imai, Chem. Lett., 1975, 891 CrossRef CAS;
(b) Y. Takeyama, K. Nozaki, K. Matsumoto, K. Oshima and K. Utimoto, Bull. Chem. Soc. Jpn., 1991, 64, 1461 CrossRef CAS;
(c) Z. Nevárez and K. A. Woerpel, Org. Lett., 2007, 9, 3773 CrossRef PubMed;
(d) R. Shintani, K. Moriya and T. Hayashi, J. Am. Chem. Soc., 2011, 133, 16440 CrossRef CAS PubMed;
(e) R. Shintani, K. Moriya and T. Hayashi, Org. Lett., 2012, 14, 2902 CrossRef CAS PubMed;
(f) J. Liu, Q. Zhang, P. Li, Z. Qu, S. Sun, Y. Ma, D. Su, Y. Zong and J. Zhang, Eur. J. Inorg. Chem., 2014, 3435 CrossRef CAS PubMed;
(g) H. Saso and W. Ando, Chem. Lett., 1988, 1567 CrossRef CAS;
(h) H. Saso, W. Ando and K. Ueno, Tetrahedron, 1989, 45, 1929 CrossRef CAS;
(i) K. M. Buchner and K. A. Woerpel, Organometallics, 2010, 29, 1661 CrossRef CAS PubMed;
(j) J. Liu, X. Sun, M. Miyazaki, L. Liu, C. Wang and Z. Xi, J. Org. Chem., 2007, 72, 3137 CrossRef CAS PubMed;
(k) N. Agenet, J.-H. Mirebeau, M. Petit, R. Thouvenot, V. Gandon, M. Malacria and C. Aubert, Organometallics, 2007, 26, 819 CrossRef CAS.
- For a review:
(a) K. Motokura and T. Baba, Green Chem., 2012, 14, 565 RSC ; For examples: ;
(b) N. Asao, E. Yoshikawa and Y. Yamamoto, J. Org. Chem., 1996, 61, 4874 CrossRef CAS;
(c) E. Yoshikawa, V. Gevorgyan, N. Asao and Y. Yamamoto, J. Am. Chem. Soc., 1997, 119, 6781 CrossRef CAS;
(d) T. Matsuda, S. Kadowaki, Y. Yamaguchi and M. Murakami, Chem. Commun., 2008, 2744 RSC;
(e) K. Motokura, S. Matsunaga, A. Miyaji, T. Yashima and T. Baba, Tetrahedron Lett., 2011, 52, 6687 CrossRef CAS PubMed;
(f) N. Asao, T. Shimada and Y. Yamamoto, J. Am. Chem. Soc., 1999, 121, 3797 CrossRef CAS;
(g) N. Asao, K. Nabatame and Y. Yamamoto, Chem. Lett., 2001, 982 CrossRef CAS;
(h) N. Asao, T. Shimada, T. Shimada and Y. Yamamoto, J. Am. Chem. Soc., 2001, 123, 10899 CrossRef CAS PubMed;
(i) T. Matsuda, Y. Yamaguchi, M. Shigeno, S. Sato and M. Murakami, Chem. Commun., 2011, 47, 8697 RSC;
(j) E. Yoshikawa, M. Kasahara, N. Asao and Y. Yamamoto, Tetrahedron Lett., 2000, 41, 4499 CrossRef CAS.
- For a radical process: K. Miura, H. Saito, T. Nakagawa, T. Hondo, J. Tateiwa, M. Sonoda and A. Hosomi, J. Org. Chem., 1998, 63, 5740 CrossRef CAS.
- For a ruthenium-catalyzed intramolecular process: S. Liu, J. Zhao, L. Kaminsky, R. J. Wilson, M. Marino and D. A. Clark, Org. Lett., 2014, 16, 4456 CrossRef CAS PubMed.
-
(a) H.-J. Zhang, P. Becker, H. Huang, R. Pirwerdjan, F.-F. Pan and C. Bolm, Adv. Synth. Catal., 2012, 354, 2157 CrossRef CAS PubMed;
(b) P. Becker, D. L. Priebbenow, H.-J. Zhang, R. Pirwerdjan and C. Bolm, J. Org. Chem., 2014, 79, 814 CrossRef CAS PubMed.
- N. Chatani, T. Takeyasu, N. Horiuchi and T. Hanafusa, J. Org. Chem., 1988, 53, 3539 CrossRef CAS.
- H. Gerhard, J. Chem. Res., Synop., 1978, 104 Search PubMed.
- For alkynylsilylation of alkynes by a three-component-coupling reaction: N. Chatani, N. Amishiro and S. Murai, J. Am. Chem. Soc., 1991, 113, 7778 CrossRef CAS.
- For recent reviews on the synthesis of enantio-enriched silicon-stereogenic organosilanes:
(a) M. Oestreich, Synlett, 2007, 1629 CrossRef CAS PubMed;
(b) A. Weickgenannt, M. Mewald and M. Oestreich, Org. Biomol. Chem., 2010, 8, 1497 RSC;
(c) L.-W. Xu, L. Li, G.-Q. Lai and J.-X. Jiang, Chem. Soc. Rev., 2011, 40, 1777 RSC;
(d) L.-W. Xu, Angew. Chem., Int. Ed., 2012, 51, 12932 CrossRef CAS PubMed;
(e) R. Shintani, Asian J. Org. Chem., 2015, 4, 510 CrossRef CAS PubMed.
-
(a) R. Shintani, H. Otomo, K. Ota and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 7305 CrossRef CAS PubMed;
(b) R. Shintani, E. E. Maciver, F. Tamakuni and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 16955 CrossRef CAS PubMed;
(c) R. Shintani, C. Takagi, T. Ito, M. Naito and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 1616 CrossRef CAS PubMed.
- For recent reviews on the rhodium-catalyzed [2+2+2] cycloaddition reactions, see:
(a) K. Tanaka, Chem. – Asian J., 2009, 4, 508 CrossRef CAS PubMed;
(b) N. Weding and M. Hapke, Chem. Soc. Rev., 2011, 40, 4525 RSC;
(c) Y. Shibata and K. Tanaka, Synthesis, 2012, 323 CAS;
(d) D. L. J. Broere and E. Ruijter, Synthesis, 2012, 2639 CAS.
- The use of 8 mol% of NaBAr4F instead of 16 mol% also provided 3a with similar efficiency.
- CCDC 1401151. See also the Electronic ESI† for details.
- See the ESI† for the optical properties of compound 5.
- For examples of catalytic asymmetric preparation of silicon-stereogenic organosilanes, see:
(a) R. J. P. Corriu and J. J. E. Moreau, Tetrahedron Lett., 1973, 14, 4469 CrossRef;
(b) T. Hayashi, K. Yamamoto and M. Kumada, Tetrahedron Lett., 1974, 15, 331 CrossRef;
(c) R. J. P. Corriu and J. J. E. Moreau, J. Organomet. Chem., 1975, 85, 19 CrossRef CAS;
(d) R. J. P. Corriu and J. J. E. Moreau, J. Organomet. Chem., 1976, 120, 337 CrossRef CAS;
(e) T. Ohta, M. Ito, A. Tsuneto and H. Takaya, J. Chem. Soc., Chem. Commun., 1994, 2525 RSC;
(f) Y. Yasutomi, H. Suematsu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 4510 CrossRef CAS PubMed;
(g) Y. Kurihara, M. Nishikawa, Y. Yamanoi and H. Nishihara, Chem. Commun., 2012, 48, 11564 RSC;
(h) K. Igawa, D. Yoshihiro, N. Ichikawa, N. Kokan and K. Tomooka, Angew. Chem., Int. Ed., 2012, 51, 12745 CrossRef CAS PubMed;
(i)
ref. 1d, e and 10; See also: ;
(j) K. Tamao, K. Nakamura, H. Ishii, S. Yamaguchi and M. Shiro, J. Am. Chem. Soc., 1996, 118, 12469 CrossRef CAS;
(k) D. R. Schmidt, S. J. O’Malley and J. L. Leighton, J. Am. Chem. Soc., 2003, 125, 1190 CrossRef CAS PubMed;
(l) K. Igawa, J. Takada, T. Shimono and K. Tomooka, J. Am. Chem. Soc., 2008, 130, 16132 CrossRef CAS PubMed;
(m) M. Onoe, K. Baba, Y. Kim, Y. Kita, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2012, 134, 19477 CrossRef CAS PubMed;
(n) X. Lu, L. Li, W. Yang, K. Jiang, K.-F. Yang, Z.-J. Zheng and L.-W. Xu, Eur. J. Org. Chem., 2013, 5814 CrossRef CAS PubMed.
-
(a) T. Hayashi, Acc. Chem. Res., 2000, 33, 354 CrossRef CAS PubMed;
(b) Y. Uozumi, A. Tanahashi, S.-Y. Lee and T. Hayashi, J. Org. Chem., 1993, 58, 1945 CrossRef CAS.
-
(a) P. von Matt and A. Pfaltz, Angew. Chem., Int. Ed. Engl., 1993, 32, 566 CrossRef PubMed;
(b) G. J. Dawson, C. G. Frost, J. M. Williams and S. J. Coote, Tetrahedron Lett., 1993, 34, 3149 CrossRef CAS.
- For example of P-(η2-arene) chelating coordination of a phosphoramidite ligand to rhodium, see: I. S. Mikhel, H. Rüegger, P. Butti, F. Camponovo, D. Huber and A. Mezzetti, Organometallics, 2008, 27, 2937 CrossRef CAS.
-
(a) Y. H. Choi, J. Y. Choi, H. Y. Yang and Y. H. Kim, Tetrahedron: Asymmetry, 2002, 13, 801 CrossRef CAS;
(b) B. M. Trost, J. P. Stambuli, S. M. Silverman and U. Schwörer, J. Am. Chem. Soc., 2006, 128, 13328 CrossRef CAS PubMed.
- M. Fañanás-Mastral, M. Pérez, P. H. Bos, A. Rudolph, S. R. Harutyunyan and B. L. Feringa, Angew. Chem., Int. Ed., 2012, 51, 1922 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures. CCDC 1401151. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc04172d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence