
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnti-Markovnikov hydroimination of terminal alkynes in gold-catalyzed pyridine construction from ammonia†
Liliang
Wang‡
a,
Lingbing
Kong‡
a,
Yongxin
Li
b,
Rakesh
Ganguly
b and
Rei
Kinjo
*a
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Nanyang Link 21, Singapore 637371, Singapore. E-mail: rkinjo@ntu.edu.sg
bNTU-CBC Crystallography Facility, Nanyang Technological University, Nanyang Link 21, Singapore 637371, Singapore
First published on 24th June 2015
Abstract
Gold-catalyzed hydroimination of terminal alkynes, giving rise to anti-Markovnikov adducts concomitant with unstable Markovnikov adducts is described. The elementary step can be applied for the construction of pyridine derivatives from ammonia and alkynes.
Acyclic and heterocyclic nitrogen-containing skeletons are ubiquitous in a myriad of naturally occurring compounds as well as in industrial products involving agrochemicals, pharmaceuticals, cosmetics, and fine chemicals.1 Hence, the development of efficient methodologies for the formation of carbon–nitrogen bonds has been a subject of considerable interest in synthetic chemistry. In recent years, metal-catalyzed hydroamination, in which C–N bonds are formed by direct addition of a nitrogen–hydrogen bond across carbon–carbon multiple bonds, has been represented as a powerful atom-economic tool for the synthesis of nitrogen-containing compounds.2 However, control of the regioselectivity is a major problem to be addressed. Thus, as classical textbooks state that the proton forms a bond with the carbon bearing fewer substituents in accordance with Markovnikov's rule, archetypal hydroamination products are also dominated by Markovnikov adducts with a branched skeleton.3 Accordingly, the preparation of nitrogen-containing compounds with a linear skeleton by direct anti-Markovnikov hydroamination remains highly challenging.4
Since the pioneering work of Beller and co-workers on the first metal-catalyzed anti-Markovnikov hydroamination of olefins in 1999,5 various methodologies for intermolecular anti-Markovnikov hydroamination using metal catalysts involving alkali metals,6 alkaline earth metals,7 organolanthanide,8 Ti,9 Re,10 Ru,11 Rh,12 Pd,13 Cu,14 Au15 have been reported to date. Although such seminal approaches have led to solid progress, these strategies often involve disadvantages such as limiting the substrate scope, harsh reaction conditions, use of strong bases, or stepwise indirect processes.
In marked contrast to recent advances in the field of hydroamination, the addition of an N–H bond of primary imines HN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR2 to alkenes and alkynes, namely hydroimination, has rarely been achieved thus far despite the significance of the predicted products, 2-aza-1,3-dienes, as synthetic intermediates for N-heterocycles.16 The use of primary imines as nucleophilic substrates is hampered by several difficulties, mainly because of their propensity to behave as electrophiles rather than nucleophiles due to the polarized CN bonds, especially in the presence of Brønsted/Lewis acids. Primary imines are known to act as directing groups by coordinating to metals which induces intramolecular C–H activation.17 In addition, oxidative homo coupling of primary imines also occurs to form an N–N bond in the presence of metals.18 To prevent such undesired processes, hydroimination has mainly been limited to intramolecular reactions with the geometrically pre-organized substrates (Scheme 1).19 Very recently, Zhao et al. first reported nickel-catalyzed intermolecular coupling between internal alkynes and aromatic N–H ketimines.20 However, employment of terminal alkynes in their system led to alkyne oligomerization instead of the desired hydroimination. Thus, to the best of our knowledge, hydroimination of terminal alkynes is still unknown to date.
CR2 to alkenes and alkynes, namely hydroimination, has rarely been achieved thus far despite the significance of the predicted products, 2-aza-1,3-dienes, as synthetic intermediates for N-heterocycles.16 The use of primary imines as nucleophilic substrates is hampered by several difficulties, mainly because of their propensity to behave as electrophiles rather than nucleophiles due to the polarized CN bonds, especially in the presence of Brønsted/Lewis acids. Primary imines are known to act as directing groups by coordinating to metals which induces intramolecular C–H activation.17 In addition, oxidative homo coupling of primary imines also occurs to form an N–N bond in the presence of metals.18 To prevent such undesired processes, hydroimination has mainly been limited to intramolecular reactions with the geometrically pre-organized substrates (Scheme 1).19 Very recently, Zhao et al. first reported nickel-catalyzed intermolecular coupling between internal alkynes and aromatic N–H ketimines.20 However, employment of terminal alkynes in their system led to alkyne oligomerization instead of the desired hydroimination. Thus, to the best of our knowledge, hydroimination of terminal alkynes is still unknown to date.
 | ||
| Scheme 1 Metal-catalyzed intramolecular and intermolecular hydroimination. | ||
Given the importance of C–N bonds in synthetic chemistry, the development of general means for the selective formation is imperative. Here, we report gold-catalyzed hydroimination of terminal alkynes, which afforded both anti-Markovnikov and Markovnikov products. We also describe the first example for construction of pyridine derivatives from ammonia and alkynes.
Our protocol for intermolecular hydroimination of terminal alkynes is based on gold catalysis employing primary ketimines HNCR2. We envisaged that (a) the soft π-acidity of gold allows interaction with alkynes effectively prior to ketimines, and (b) the presence of two R-substituents at the imine carbon of ketimines suppresses the attack by nucleophiles kinetically. We also reasoned that incorporation of a bulky ligand into gold might minimise the interaction with ketimines by steric repulsion, which will induce selective activation of terminal alkynes rather than the undesired reaction pathway. Among the various ligands available, pyrid-2-ylidene ligands are considered to be good candidates because substitutions at 1- and 3-positions maximize the steric impact at the gold center due to the six-membered ring skeleton.21 Pyrid-2-ylidenes are also recognized as strong σ-donor ligands, which will contribute to promote substrate exchange, necessary for a high turnover in catalysis, as well as the stability of the complex.22 Hence, to commence our studies, we designed a novel gold chloride complex supported by a pyrid-2-ylidene ligand L bearing a 1,3,5-trimethylphenyl group and 3,5-di-tert-butylphenyl groups. When the deprotonation of a pyridinium salt [L–H]+[CF3SO3]− was performed with lithium hexamethyldisilazide (LiHMDS)(3 eq.), which is followed by the addition of chloro(tetrahydrothiophene)gold(I), a clean reaction occurred, showing a characteristic signal for the carbene carbon at 187.8 ppm in the 13C NMR spectrum. After the products were worked up, LAuCl was obtained as a white solid in 49% yield (Scheme 2). LAuCl was fully characterized using standard spectroscopic methods, including a single crystal X-ray diffraction study.23 LAuCl is air-stable and can be stored for several months without significant decomposition, both in solution and in the solid state (m.p.; 220 °C).
 | ||
| Scheme 2 Synthesis of the gold complex LAuCl. | ||
With the precatalyst LAuCl in hand, we next examined its catalytic activity in the hydroimination of terminal alkynes. Recently, Toste and co-workers reported that the gold-catalyzed reaction of phenylacetylene 1a with N,1-diphenylmethanimine PhNCHPh afforded propargylamine.24 In marked contrast, the reaction between the two equivalents of 1a and benzophenone imine 2a with a catalytic amount of LAuCl produced a mixture of (Z)-1,1-diphenyl-N-styrylmethanimine 3a and 1,1-diphenyl-N-(1-phenylvinyl) methanimine 4a after 1 hour at 150 °C, and unexpectedly, the yield of the anti-Markovnikov adduct 3a was nearly identical to that of Markovnikov adduct 4a (3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a ≈ 1:1). We also observed that 4a gradually decomposed to an unidentified mixture under the reaction conditions (see the ESI†). Therefore, in order to substantiate the apparent regioselectivity, the reaction was repeated and monitored by NMR spectroscopy. Under the screening reaction conditions, the highest production of the anti-Markovnikov adduct 3a (51%) was reported after 6 hours (Table 1, entry 1).
:4a ≈ 1:1). We also observed that 4a gradually decomposed to an unidentified mixture under the reaction conditions (see the ESI†). Therefore, in order to substantiate the apparent regioselectivity, the reaction was repeated and monitored by NMR spectroscopy. Under the screening reaction conditions, the highest production of the anti-Markovnikov adduct 3a (51%) was reported after 6 hours (Table 1, entry 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 | 2 | T (°C) | Time (h) | Yieldb,c (3) (%) | Product ratiob (3:4) |
| a Reaction conditions: 1 (0.4 mmol), 2 (0.2 mmol), LAuCl (5 mol%) and KB(C6F5)4 (5 mol%), C6D6 (0.5 mL), 150 °C. b Yields and selectivity were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as the internal standard. c Isolated yields are given in parentheses. | ||||||
| 1 | 1a (R = H) |

|
150 | 6 | 51(47) |
3a:4a = 6.2:1 |
| 2 | 1b (R = Br) | 150 | 6 | 54(50) |
3b:4b = 100:0 |
|
| 3 | 1c (R = Me) | 150 | 6 | 43(39) |
3c:4c = 100:0 |
|
| 4 | 1a (R = H) |
|
150 | 6 | 40(37) |
3d:4d = 100 :0 |
| 5 | 1b (R = Br) | 150 | 6 | 48(44) |
3e:4e = 100:0 |
|
| 6 | 1c (R = Me) | 150 | 6 | 42(37) |
3f:4f = 100:0 |
|
| 7 | 1a (R = H) |

|
150 | 5 | 50(47) |
3g:4g = 100:0 |
| 8 | 1b (R = Br) | 150 | 6 | 48(42) |
3h:4h = 100:0 |
|
| 9 | 1c (R = Me) | 150 | 5 | 36(32) |
3i:4i = 100:0 |
|
| 10 | 1a (R = H) |
|
150 | 4 | 51(47) |
3j:4j = 4.3:1 |
| 11 | 1b (R = Br) | 150 | 3 | 55(51) |
3k:4k = 6.1 :1 |
|
| 12 | 1c (R = Me) | 150 | 3 | 43(39) |
31:41 = 6.1 :1 |
|
| 13 | 1a (R = H) |

|
150 | 5 | — |

|
To further probe the formation of an anti-Markovnikov product, we performed a 13C-labeling experiment with a 13C-labeled phenylacetylene 1a*, which decisively afforded 3a* (Fig. 1a). Control reactions revealed the innocence of potassium tetrakis(pentafluorophenyl)borate KB(C6F5)4, demonstrating the essential role of the gold complex LAuCl in this reaction. In fact, the reaction shut down in the absence of the Au precatalyst. With AgOTf instead of KB(C6F5)4 under similar reaction conditions, the product 3a was obtained in a lower yield (20%). When 1-bromo-4-ethynylbenezene 1b was used (entry 2), the formation of anti-Markovnikov product 3b was observed prior to Markovnikov product 4b even in the early stage of the reaction (after 1 h; 3b:4b = 12:1), and the highest yield (54%) of 3b was obtained after 6 hours when 4b decomposed completely. Treatment of 1a and 4-ethynyltoluene 1c afforded a similar result in which anti-Markovnikov adduct 3c was formed in 43% yield after 6 hours (entry 3). These results indicate that the apparent anti-Markovnikov regioselectivity is concomitant with the decomposition of Markovnikov products in addition to the formation of a mixture of other unidentified products, whereas the stability of anti-Markovnikov products encouraged us for further exploration. Reaction of 2a with an internal alkyne, diphenylacetylene, was also tested, which afforded N-(1,2-diphenylvinyl)-1,1-diphenylmethanimine in 78% yield after 22 h (see the ESI†).
 | ||
| Fig. 1 (a) Representations of 13C-labeled phenylacetylene 1a*, (Z)-1,1-diphenyl-N-styrylmethanimine 3a*, and 2-benzyl-4,6-diphenylpyridine 5a*. (b) Proposed reaction pathway for the construction of pyridine 5 from alkynes 1 and ammonia. | ||
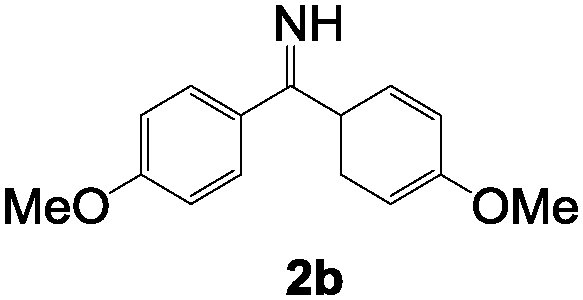
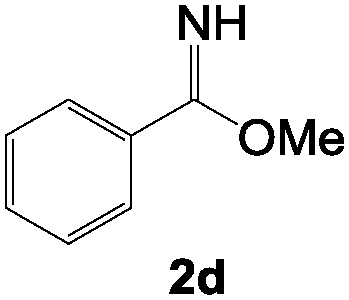
To test the scope of the hydroimination with respect to imines, we employed various imine substrates. Each reaction was monitored using NMR spectroscopy, and the results for the anti-Markovnikov adduct 3 in the highest yield are summarized in Table 1. Standard functional groups are tolerated, including diaryl imines featuring p-methylphenyl groups 2b, p-fluorophenyl groups 2c, as well as methyl benzimidate 2d. All imine substrates 2a–d examined in this study reacted well with alkynes 1a–c and afforded 2-aza-1,3-dienes 3a–l in moderate yields. Note that catalytic formation of 2-aza-1,3-dienes such as 3 and 4 from terminal alkynes and imines has never been reported before. It is also noteworthy to mention that under similar conditions, employment of other gold catalysts such as (Ph3P)AuCl and (IPr)AuCl afforded no and a few (<10%) products, respectively. We also examined the reaction of bis-aliphatic imine, 2,2,4,4-tetramethylpentan-3-imine (tBu2CNH) with 1a, which gave a complex mixture.
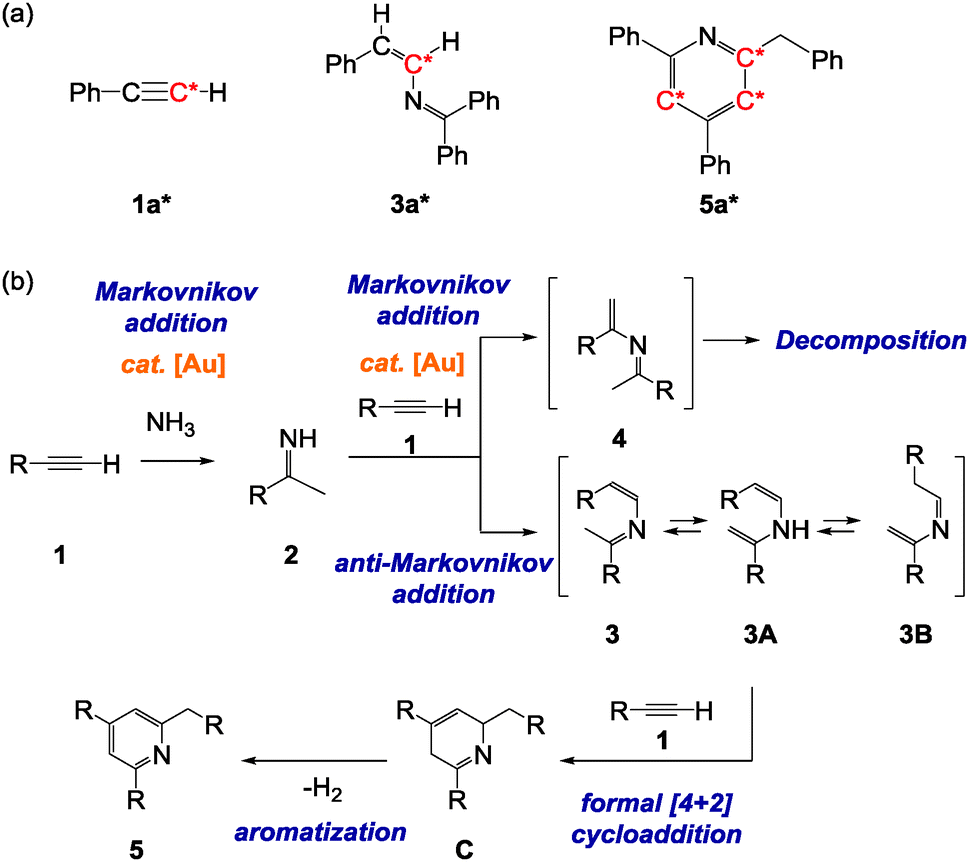
Reaction of 1a with 1-phenylethan-1-imine 2e also proceeded under similar reaction conditions. However, neither 3m nor 4m was detected. Instead, only unidentified self-decomposed products of 2e were observed after the reaction. To our surprise, when a large excess of 1a was used, we obtained 2-benzyl-4,6-diphenylpyridine 5 in 51% yield after 5 hours, indicating that two equivalents of 1a were involved in the reaction (Table 1, entry 13). Although we attempted to confirm the reaction intermediates by varying the reaction temperature, time, and substrate ratio, 5a was the only detectable product under any condition.25 Presumably, the instability of the corresponding 2-aza-1,3-diene intermediate caused fast cyclization with a second alkyne. The high reactivity of the intermediates may be due to the presence of the sterically less demanding methyl group at the imine carbon, which also could induce tautomerization to transient enamine intermediates. It has already been shown that less-hindered 2-aza-1,3-diene derivatives react with unsaturated molecules to generate cyclic products.16,26
Bertrand and co-workers have reported that a catalytic amount of cyclic (alkyl)(amino)carbene gold complexes effectively promotes the addition of ammonia (NH3) across alkynes and allenes.27 In their study, hydroamination of a terminal arylalkyne, 4-ethynyltoluene, proceeded with Markovnikov regioselectivity, which afforded 1-arylethan-1-imine. On the basis of these results, we attempted the direct synthesis of pyridine skeletons, common components of natural products and pharmaceuticals,28 from alkynes and NH3 through the anti-Markovnikov hydroimination–cyclization sequence. We postulate that treatment of terminal arylalkynes 1 and NH3 in the presence of our gold catalyst LAuCl also would generate 1-arylethan-1-imines 2via Markovnikov hydroamination rather than anti-Markovnikov selectivity due to the less bulkiness of the ammonia molecules. The imines formed in situ would further react with a second alkyne in an anti-Markovnikov fashion to give 2-aza-1,3-diene intermediates 3 which would isomerize to 3A and 3B followed by cyclization with an additional alkyne. Finally, dehydrogenative aromatization from intermediates C would afford pyridine derivatives 5 (Fig. 1b).
To bear out this hypothesis, 2.5 equivalents of 1a were treated with NH3 in the presence of the gold complex LAuCl (1 mol%). To our delight, after 12 hours at 150 °C, 5a was obtained in 43% yield (Table 2a, entry 1). Interestingly, the spontaneous aromatization by dehydrogenation was induced even without an oxidant. To gain insight into the reaction pathway, we performed further experiments. Reaction of 1a with a large excess of NH3 exclusively afforded 1-phenylethan-1-imine 2e, confirming that the initial step is a Markovnikov hydroamination of alkyne, affording an enamine which may subsequently tautomerize to imine 2e. Next, a 13C-labeling experiment was conducted with 1a*. When 1a* was employed under the same reaction conditions, 5a* was produced which supports the proposed reaction pathway (Fig. 1a). The scope of the catalytic reaction was briefly examined for a variety of alkynes 1 (Table 2a). Terminal alkynes with electron-donating as well as electron-withdrawing aromatic groups were well tolerated (Table 2a, entries 2–7, 9, 10). The relatively low yield for 1h was probably due to the adoption of the extremely strong electron-withdrawing CF3-group (Table 2a, entry 8). 2-Ethynylthiophene and 3-ethynylthiophene also exhibited tolerance to the reaction conditions (Table 2a, entries 11 and 12). Finally, our preliminary test showed that this strategy could be extended to a three-component coupling reaction employing two different terminal alkynes and NH3, which afforded rather complex pyridine derivatives 6 (Table 2b). This result illustrates the potential for application of the preparation of various heterocycles, although co-products 5 assembled from the mono-component alkyne were also formed in this reaction.
| (a) | |||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| Entry | Substrate | 5 Yieldb,c | Entry | Substrate | 5 Yieldb,c | ||
| a Reaction conditions: 1 (1 mmol), NH3 (0.25 mmol), LAuCl (1 mol%) and KB(C6F5)4 (1 mol%), C6D6 (0.7 mL), 150 °C. b Yields and selectivity were determined by 1H NMR spectroscopy using 1,4-di-t-butylbenzene as the internal standard. c Isolated yields are given in parentheses. d For other examples, see the ESI (Scheme S4). | |||||||
| 1 | 1a |
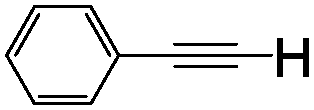
|
5a 43% (40%) | 7 | 1g |

|
5g 41% (38%) |
| 2 | 1b |

|
5b 38% (34%) | 8 | 1h |

|
5h 23% (19%) |
| 3 | 1c |
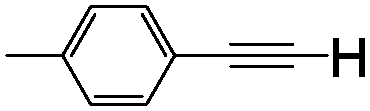
|
5c 33% (30%) | 9 | 1i |

|
5i 30% (28%) |
| 4 | 1d |

|
5d 31% (27%) | 10 | 1j |
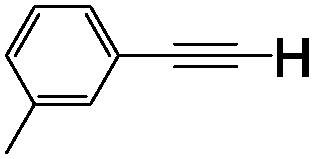
|
5j 41% (37%) |
| 5 | 1e |

|
5e 31% (28%) | 11 | 1k |

|
5k 20% (17%) |
| 6 | 1f |

|
5f 45% (43%) | 12 | 1l |
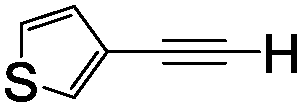
|
51 48% (45%) |

| (b)d |
|---|

|
To investigate the reaction mechanism, we tested the reaction of 3a with 1c in the presence of LAuCl/KB(C6F5)4 (5 mol%) under similar reaction conditions. However, products corresponding to C were not detected, and only a complex mixture was obtained. We postulated that a Me-group at the imine carbon in 3 is necessary for the formal [4+2] cycloaddition because it could isomerize to transient 3B, to which alkyne 1 readily approaches due to the less steric hindrance. Meanwhile, it has been reported that the formal [4+2] cycloaddition between azadienes and unsaturated compounds proceeds without any catalysts.29 Further study of the relevant gold catalysis30 with LAuCl is currently under investigation.
The authors gratefully acknowledge financial support from Nanyang Technological University and PSF/A*STAR (SERC 1321202066) of Singapore.
Notes and references
- (a) S. A. Lawrence, Amines: Synthesis, Properties, and Applications, Cambridge University Press, New York, 2004 Search PubMed; (b) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435 RSC.
- (a) A. L. Reznichenko and K. C. Hultzsch, Top. Organomet. Chem., 2013, 43, 51 CrossRef CAS; (b) J. F. Hartwig, Organotransition Metal Chemistry, University Science Books, Mill Valley, CA, 2010, ch. 16.5, p.700 Search PubMed; (c) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef PubMed; (d) R. Severin and S. Doye, Chem. Soc. Rev., 2007, 36, 1407 RSC; (e) E. Mizushima, T. Hayashi and M. Tanaka, Org. Lett., 2003, 5, 3349 CrossRef CAS PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry, Wiley, New York, 5 edn, 2001 Search PubMed.
- J. Haggin, Chem. Eng. News, 1993, 71, 23 Search PubMed.
- M. Beller, H. Trauthwein, M. Eichberger, C. Breindl, J. Herwig, T. E. Müller and O. R. Thiel, Chem. – Eur. J., 1999, 5, 1306 CrossRef CAS.
- (a) P. Horrillo-Martínez, K. C. Hultzsch, A. Gil and V. Branchadell, Eur. J. Org. Chem., 2007, 3311 CrossRef PubMed; (b) K. Kumar, D. Michalik, I. Garcia Castra, A. Tillack, A. Zapf, M. Arlt, T. Heinrich, H. Böttcher and M. Beller, Chem. – Eur. J., 2004, 10, 746 CrossRef CAS PubMed.
- (a) C. Brinkmann, A. G. M. Barrett, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2012, 134, 2193 CrossRef CAS PubMed; (b) A. G. M. Barrett, C. Brinkmann, M. R. Crimmin, M. S. Hill, P. Hunt and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 12906 CrossRef CAS PubMed.
- J.-S. Ryu, G. Y. Li and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 12584 CrossRef CAS PubMed.
- A. Tillack, I. G. Castro, C. G. Hartung and M. Beller, Angew. Chem., Int. Ed., 2002, 41, 2541 CrossRef CAS.
- S. S. Yudha, Y. Kuninobu and K. Takai, Org. Lett., 2007, 9, 5609 CrossRef CAS PubMed.
- (a) M. Arndt, K. S. M. Salih, A. Fromm, L. J. Goossen, F. Menges and G. Niedner-Schatteburg, J. Am. Chem. Soc., 2011, 133, 7428 CrossRef CAS PubMed; (b) M. Utsunomiya and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 2702 CrossRef CAS PubMed.
- M. Utsunomiya, R. Kuwano, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 5608 CrossRef CAS PubMed.
- S. M. Bronner and R. H. Grubbs, Chem. Sci., 2014, 5, 101 RSC.
- R. P. Rucker, A. M. Whittaker, H. Dang and G. Lalic, J. Am. Chem. Soc., 2012, 134, 6571 CrossRef CAS PubMed.
- J. C. Timmerman, B. D. Robertson and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2015, 54, 2251 CrossRef CAS PubMed.
- F. Palacios, C. Alonso, M. Rodríguez, E. M. d. Marigorta and G. Rubiales, Eur. J. Org. Chem., 2005, 1795 CrossRef CAS PubMed.
- (a) R. He, Z.-T. Huang, Q.-Y. Zheng and C. Wang, Angew. Chem., Int. Ed., 2014, 53, 4950 CrossRef CAS PubMed; (b) S. Gupta, J. Han, Y. Kim, S. W. Lee, Y. H. Rhee and J. Park, J. Org. Chem., 2014, 79, 9094 CrossRef CAS PubMed; (c) D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2013, 52, 10630 CrossRef CAS PubMed; (d) J. Zhang, A. Ugrinov and P. Zhao, Angew. Chem., Int. Ed., 2013, 52, 6681 CrossRef CAS PubMed.
- A. Laouiti, M. M. Rammah, M. B. Rammah, J. Marrot, F. Couty and G. Evano, Org. Lett., 2012, 14, 6 CrossRef CAS PubMed.
- (a) D. Pflästerer, P. Dolbundalchok, S. Rafique, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2013, 355, 1383 CrossRef PubMed; (b) V. H. L. Wong, T. S. A. Hor and K. K. Hii, Chem. Commun., 2013, 49, 9272 RSC; (c) A. S. K. Hashmi, M. Rudolph, S. Schymura, J. Visus and W. Frey, Eur. J. Org. Chem., 2006, 4905 CrossRef CAS PubMed; (d) J.-E. Kang, H.-B. Kim, J.-W. Lee and S. Shin, Org. Lett., 2006, 8, 3537 CrossRef CAS PubMed.
- R. S. Manan, P. Kilaru and P. Zhao, J. Am. Chem. Soc., 2015, 137, 6136 CrossRef CAS PubMed.
- K. Hata, Y. Segawa and K. Itami, Chem. Commun., 2012, 48, 6642 RSC.
- (a) A. A. Tukov, A. T. Normand and M. S. Nechaev, Dalton Trans., 2009, 7015 RSC . For further examples of catalyst stabilization by bulky ligands, see: ; (b) M. C. B. Jaimes, F. Rominger, M. M. Pereira, R. M. B. Carrilho, S. A. C. Carabineiro and A. S. K. Hashmi, Chem. Commun., 2014, 50, 4937 RSC; (c) M. C. B. Jaimes, C. R. N. Böhling, J. M. Serra-Becerra and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 7963 CrossRef PubMed.
- Experimental data for [L–H]+[CF3SO3]−, LAuCl, and products 3, 5 and 6 are available in the ESI†.
- M. J. Campbell and F. D. Toste, Chem. Sci., 2011, 2, 1369 RSC.
- A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232 CrossRef CAS PubMed.
- F. Palacios, C. Alonso, C. Tobillas and G. Rubiales, Heterocycles, 2004, 64, 229 CrossRef CAS PubMed.
- V. Lavallo, G. D. Frey, B. Donnadieu, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 5224 CrossRef CAS PubMed.
- A. Brossi, in The Alkaloids: Chemistry and Pharmacology, ed. G. A. Cordell, Academic Press, San Diego, 1993, vol. 43, p.119 Search PubMed.
- (a) S. Jayakumar, M. P. S. Ishar and M. P. Mahajan, Tetrahedron, 2002, 58, 379 CrossRef CAS; (b) S. M. Weinreb and P. M. Scola, Chem. Rev., 1989, 89, 1525 CrossRef CAS.
- (a) A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS PubMed; (b) A. Arcadi, Chem. Rev., 2008, 108, 3266 CrossRef CAS PubMed; (c) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and calculation details, and crystallographic information for LAuCl, 3h, 3i, 5b, 5g and 6d. CCDC 1051367–1051372. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc04091d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |