
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regular assembly of cage siloxanes by hydrogen bonding of dimethylsilanol groups†
Naoto
Sato
a,
Yoshiyuki
Kuroda
b,
Takuya
Abe
a,
Hiroaki
Wada
a,
Atsushi
Shimojima
*a and
Kazuyuki
Kuroda
*ac
aDepartment of Applied Chemistry, Faculty of Science and Engineering, Waseda University, 3-4-1 Ohkubo, Shinjuku-ku, Tokyo 169-8555, Japan
bWaseda Institute for Advanced Study, Waseda University, Tokyo 169-8050, Japan
cKagami Memorial Research Institute for Materials Science and Technology, Waseda University, 2-8-26 Nishiwaseda, Shinjuku-ku, Tokyo 169-0051, Japan. E-mail: shimojima@waseda.jp; kuroda@waseda.jp; Fax: +81-3-5286-3199
First published on 3rd June 2015
Abstract
A new class of ordered silica-based materials has been prepared by hydrogen bond-directed assembly of cage siloxanes modified with dimethylsilanol groups, providing a soft-chemical approach to crystalline silica materials with molecularly designed architectures.
Crystalline silica-based materials such as zeolites and layered silicates are important for applications in catalysis, adsorption and separation.1 These materials are generally synthesized under hydrothermal conditions; however, the complex crystallization processes involving dissolution and redeposition of silica make it difficult to design their structures at a molecular level. Controlled assembly of well-defined building blocks is expected to provide a solution to this problem. Cage-type siloxane oligomers are useful as building blocks of siloxane-based nanomaterials because of the rigid framework and ease in modification at the cage corners.2 Siloxane cages are also known as secondary building units of zeolite frameworks. However, regular arrangement of the siloxane cages by soft chemical processes still remains a challenge. Although a certain progress has been made by amphiphilic assembly of cage siloxanes bearing either hydrophobic or hydrophilic chain3 and by cross-linking of cage siloxanes with rigid organic groups,4 truly crystalline materials have never been reported so far.
Various supramolecular assemblies can be constructed by hydrogen bonding between designed organic molecules.5 Modification of Si-based molecules, including alkoxysilanes and cage siloxanes, with organic groups capable of hydrogen bonding is a straightforward approach to achieve ordered assembly.6,7 Utilization of silanol (Si–OH) groups is also interesting because of the ability to form both hydrogen bonds and siloxane bonds. Formation of stable molecular crystals stabilized by hydrogen bonding networks was reported for bulky organosilanols.8 Furthermore, Kawakami et al. reported the synthesis of crystalline materials where the cage siloxanes with eight organosilanol groups are hydrogen bonded to form three-dimensional (3D) frameworks containing solvent molecules.9 To extend such organosilane chemistry for constructing ordered siloxane frameworks, we synthesized a cage siloxane modified with diphenylsilanol groups attached at the cage corner by siloxane bonds.10 This molecule underwent solid-state condensation by thermal treatment; however, structural regularities both before and after the heat treatment were very low. It is likely that the bulky diphenylsilyl groups attached close to the cage corners sterically hinder the formation of ordered hydrogen bonding networks.
Here we report the formation of highly ordered layered crystals from cage siloxane modified with dimethylsilanol (–SiMe2OH) groups (1 in Fig. 1). The anisotropic, two-dimensional assembly of the cubic-shaped cage siloxane molecules is an interesting phenomenon that is different from the aforementioned 3D assembly of organosilanol-modified cage siloxanes. Despite the simple structure of 1, having no bulky organic groups, the silanol groups are stabilized in the crystals by forming hydrogen-bonding networks. Unique structural changes were observed upon drying and/or treatment with hydrophobic organic molecules. Furthermore, the thermal treatment of the crystals induced solid-state polycondensation to form an ordered siloxane structure.
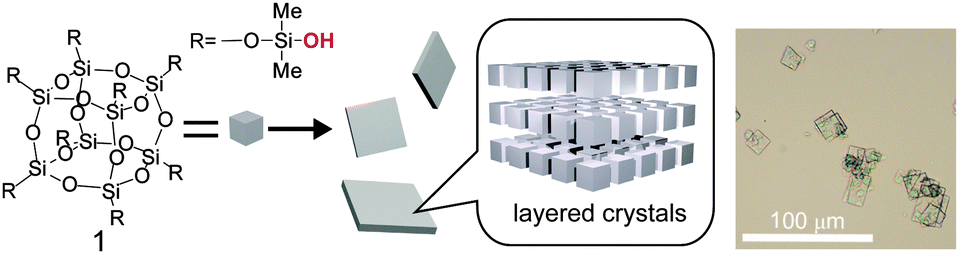 | ||
| Fig. 1 A scheme for the formation of layered crystals from cage siloxane modified with dimethylsilanol groups (1). The optical microscopy image shows as-deposited crystals. | ||
The dimethylsilanol-modified cubic siloxane (Si8O12(OSiMe2OH)8, 1) was synthesized by silylation of cubic octameric silicate ions (Si8O208−) with chlorodimethylsilane,11 followed by conversion of the SiH groups into SiOH groups using THF as a solvent and Pd/C as a catalyst (see ESI† for details). The crude crystals of 1, deposited by adding the THF solution into hexane, were recrystallized from 2-methyltetrahydrofuran and toluene. The square or rectangular-shaped crystalline precipitates (Fig. 1 right) were recovered by filtration and air-dried at ca. 25 °C to yield white powders. Although the synthesis of 1 was reported in the literature,12 crystallization of 1 has not been studied previously.
The crystals of 1 were easily dissolved in THF even after drying at 25 °C for several days, though a small amount of undissolved species remained. The 29Si NMR and mass analyses showed that the silanol groups of 1 remained intact without condensation (Fig. S1 and S2 in ESI†). The FT-IR spectrum of the crystals showed a broad, strong band centered at 3300 cm−1 which can be attributed to hydrogen-bonded silanol groups, while the band due to free silanol groups (ca. 3700 cm−1) was negligible (Fig. S3(a) in ESI†). The band attributed to silanol groups was also observed at 884 cm−1.13 A thin platelike morphology was observed by a field-emission scanning electron microscope (FE-SEM) (Fig. 2(a)). A unique laminated morphology was observed for the cross-section of the fractured plate (Fig. 2(b)).
 | ||
| Fig. 2 FE-SEM images of the crystals of 1: (a) low magnification image from the surface and (b) high magnification image of the fractured cross-section. | ||
Stepwise structural changes of the crystals were observed during drying. The X-ray diffraction (XRD) pattern of the wet crystals before drying exhibited the most intense peak at d = 1.57 nm, and similar intense peaks of dried samples appeared at 1.40 nm and finally at 1.11 nm (Fig. 3(a)). This shift suggests that the as-filtered crystals contain the solvent molecules in the framework, and the structural change occurs upon their removal, as discussed later. Although the crystal structure has not been clarified yet because of the difficulty in obtaining a large single crystal required for single-crystal X-ray analysis, the platelike morphology as well as the cleavage planes observed for the cross-section of the plate (Fig. 2(b)) strongly suggest that the cage siloxanes (1) are regularly arranged to form a lamellar structure by hydrogen bonding of dimethylsilanol groups. The most intense peaks at the lowest 2θ region should correspond to the lamellar periodicity (for details, see Fig. S4 and the following discussion in ESI†). From the molecular size of 1 (ca. 1.2 nm), it is reasonable to consider that the each layer is composed of a monolayer of the cages. The electron diffraction (ED) pattern of the plate (Fig. 3(b)) shows the structural periodicity in the in-plane direction. The appearance of square arrays of distinct spots instead of a ring pattern indicates that the plate is monocrystalline and that the cage siloxanes are arranged in a lattice pattern.
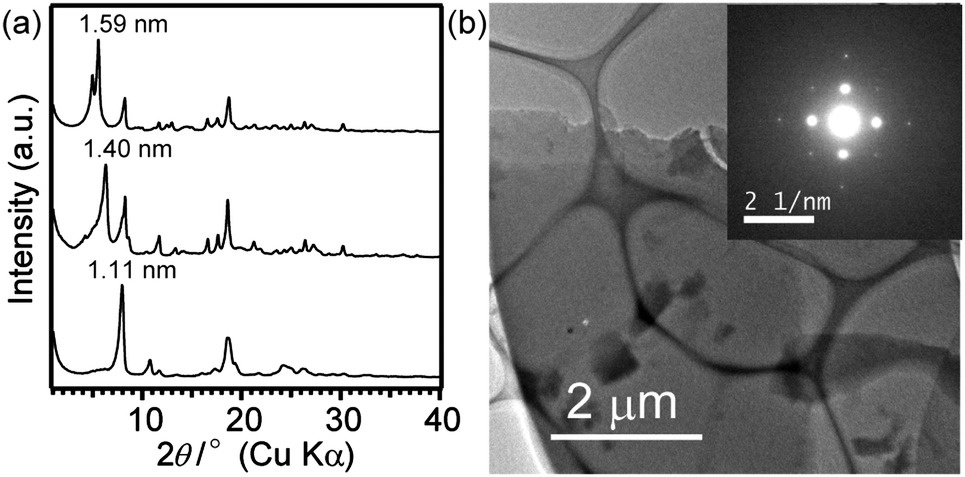 | ||
| Fig. 3 (a) Powder XRD patterns of the crystals of 1 before drying (top), after drying for 1 h (middle) and 10 h (bottom); (b) TEM image of a fully dried crystal of 1 on a carbon-coated micro-grid (inset: the ED pattern). | ||
The formation of the low-dimensional, layered structures from highly symmetric cage siloxanes is probably due to the flexible siloxane bond between the dimethylsilanol groups and the corner Si atoms of the cage siloxane. In previously reported hydrogen bonding networks of cage siloxanes,9 rigid organic spacers define the direction of the silanol groups to yield 3D structures. In the present study, possible rotation of the Si–O–Si linkages should allow formation of more diverse structures. When the cage siloxanes are arranged in a square lattice, four silanol groups can form a ring of intermolecular hydrogen bonds8 at each corner of the cage siloxane (Fig. S5 in ESI†). In this case, the molecular modelling confirmed that hydrogen bonds cannot form between the adjacent layers because two methyl groups stand toward interlayer direction.
We recently studied hydrolysis and polycondensation of a cage siloxane possessing eight dimethylethoxy groups (Si8O12(OSiMe2OEt)8) under acidic conditions. The resulting xerogel showed only a broad XRD peak,14 similar to many of the conventional materials prepared from cage siloxanes. This is due to the unstability of dimethylsilanol groups formed by hydrolysis of the SiOEt groups. A small amount of acid or base, generally added as a hydrolysis catalyst for alkoxy groups, promotes condensation of silanol groups as well, which is undesirable for the formation of ordered molecular assemblies from hydrolyzed species. Introduction of bulky diphenylsilyl groups was therefore required to stabilize the silanol groups as we reported previously.10 In contrast, using a Pd/C catalyst in the present study allowed conversion of the SiH groups into silanol groups under neutral conditions. This is the key for the stabilization of the silanol groups without condensation. Thus, this method is effective for the synthesis of the cage siloxane possessing stable silanol groups.
The involvement of the organic solvents in the structural change of the crystals was investigated. By carefully controlling the degree of drying, the peak at d = 1.39 nm can be observed predominantly (Fig. S6(a) in ESI†). At this stage, the presence of toluene molecules was confirmed by NMR analysis (Fig. S7 in ESI†). An increase of the d-spacing from 1.39 nm to 1.62 nm occurred when this sample was dispersed in 1,3,5-trimethylbenzene (TMB) (Fig. S6(b) in ESI†). Such an increase in the d spacing was not observed for the fully dried sample showing the peak at d = 1.11 nm. These results imply that the interlayer toluene molecules were replaced with trimethylbenzene molecules. Note that the shift was not attributed to dissolution–redeposition because the platelike morphology was preserved. The structural change of the hydrogen-bonded network of cage siloxanes triggered by organic solvent is reported here for the first time.
Although 1 in the crystals was stable under ambient conditions, polycondensation of the silanol groups occurred to form siloxane networks by heat treatment. A weight loss (∼6%) was observed above 200 °C due to dehydration by polycondensation of silanol groups (Fig. S8 in ESI†). We found that condensation gradually proceeded even at 150 °C, while keeping the platelike morphology of the crystals (Fig. S9(a) and S9(b) in ESI†). The IR band of silanol groups at 884 cm−1 disappeared after the heat treatment (Fig. S3(b)). In the 29Si MAS NMR spectrum (Fig. 4(a)), the D1 signal of dimethylsilanol groups (ca. −10 ppm) decreased significantly, and a new signal assigned to the D2 unit (SiMe2(OSi)2) appeared at ca. −20 ppm. The retention of the –SiMe2– groups was confirmed by the intensity ratio of Dn/Qm (Dn: SiMe2(OH)2−n(OSi)n), (Qm: Si(OH)4−m(OSi)m). On the basis of the structural model (Fig. S5, ESI†), it is speculated that two Si–O–Si linkages are formed from the four hydrogen-bonded silanol groups to form an interconnected cage siloxane network (Fig. S10 in ESI†). The small Q3 signal (at −100 ppm) indicates partial cleavage of the Si–O–Si linkages possibly caused by the attack of Si–OH groups and/or H2O generated by condensation. Nonetheless, the high Q4/Q3 ratio suggests that the siloxane cages are mostly retained. This is also supported by the relatively narrow widths of the D2 and Q4 signals compared to those of the amorphous xerogel prepared by co-condensation of dimethyldiethoxysilane and tetraethoxysilane.13 The XRD and ED patterns (Fig. 4(b–d)) clearly show that the ordered arrangement of the siloxane cages still remains in the sample after heating. It should also be noted that the stability of the structure under electron beam irradiation has been apparently enhanced by polycondensation. The TG–DTA analysis showed that combustion of the SiMe groups occurred at >400 °C in air, indicating a high stability of the product (Fig. S11 in ESI†). The N2 adsorption–desorption isotherm (Fig. S12 in ESI†) showed that 1 after the heat treatment had a low BET surface area (21 m2 g−1). To the best of our knowledge, this is the first report on the formation of a siloxane structure with a certain crystallinity consisting of cage siloxane units.
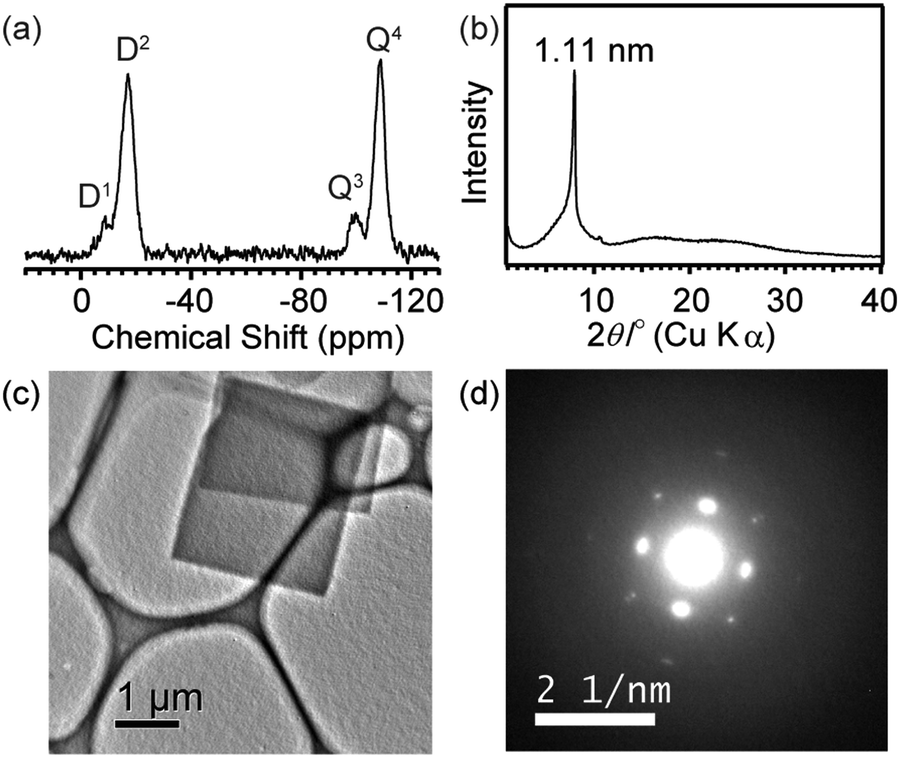 | ||
| Fig. 4 (a) Solid-state 29Si MAS NMR spectrum, (b) powder XRD pattern, (c) TEM image and (d) ED pattern of the crystals of 1 after heating at 150 °C for 8 days. | ||
In conclusion, we have demonstrated the formation of layered crystals of cage siloxane modified with SiMe2OH groups. It is suggested that cage siloxane units are assembled into a lamellar structure. The crystal was heat treated to yield siloxane network with some periodicity. This soft chemical method based on the ordered assembly of cage siloxane by hydrogen bonding will contribute to precise preparation of siloxane-based materials with various structures and morphologies by molecular design of the building blocks. One of the most important challenges is the creation of novel porous crystalline materials.
The authors are grateful to Dr K. Kawahara and Dr Y. Hagiwara for fruitful discussions. This work was supported in part by a Grant-in-Aid for Scientific Research B (No. 15H03879) from Japan Society for the Promotion of Science (JSPS) and by a Grant-in-Aid for Scientific Research on Innovative Areas “New Polymeric Materials Based on Element-Blocks (Area No. 2401)” from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
Notes and references
- (a) M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS PubMed; (b) Y. Ide, N. Ochi and M. Ogawa, Angew. Chem., Int. Ed., 2011, 50, 654 CrossRef CAS PubMed.
- (a) D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081 CrossRef CAS PubMed; (b) R. M. Laine, J. Mater. Chem., 2005, 15, 3725 RSC; (c) K. Tanaka and Y. Chujo, Polym. J., 2013, 45, 247 CrossRef CAS PubMed.
- (a) A. Shimojima, R. Goto, N. Atsumi and K. Kuroda, Chem. – Eur. J., 2008, 14, 8500 CrossRef CAS PubMed; (b) K. Y. Mya, X. Li, L. Chen and C. He, J. Nanosci. Nanotechnol., 2006, 6, 3955 CrossRef CAS PubMed.
- (a) W. Chaikittisilp, A. Sugawara, A. Shimojima and T. Okubo, Chem. Mater., 2010, 22, 4841 CrossRef CAS; (b) Y. Kim, K. Koh, M. F. Roll, R. M. Laine and A. J. Matzger, Macromolecules, 2010, 43, 6995 CrossRef CAS; (c) M. F. Roll, J. W. Kampf, Y. Kim, E. Yi and R. M. Laine, J. Am. Chem. Soc., 2010, 132, 10171 CrossRef CAS PubMed.
- J. Tian, P. K. Thallapally and B. P. McGrail, CrystEngComm, 2012, 14, 1909 RSC.
- (a) B. P. Pichon, M. Wong Chi Man, P. Dieudonné, J.-L. Bantignies, C. Bied, J.-L. Sauvajol and J. J. E. Moreau, Adv. Funct. Mater., 2007, 17, 2349 CrossRef CAS PubMed; (b) M. Fernandes, X. Cattoën, V. de Z. Bermudez and M. Wong Chi Man, CrystEngComm, 2011, 13, 1410 RSC; (c) L. D. Carlos, V. de Z. Bermudez, V. S. Amaral, S. C. Nunes, N. J. O. Silva, R. A. S. Ferreira, J. Rocha, C. V. Santilli and D. Ostrovskii, Adv. Mater., 2007, 19, 341 CrossRef CAS PubMed.
- (a) R.-S. Shih, C.-H. Lu, S.-W. Kuo and F.-C. Chang, J. Phys. Chem. C, 2010, 114, 12855 CrossRef CAS; (b) Y.-C. Wu and S.-W. Kuo, J. Mater. Chem., 2012, 22, 2982 RSC; (c) C.-C. Cheng, Y.-C. Yen, F.-H. Ko, C.-W. Chu, S.-K. Fan and F.-C. Chang, J. Mater. Chem., 2012, 22, 731 RSC.
- V. Chandrasekhar, R. Boomishankar and S. Nagendran, Chem. Rev., 2004, 104, 5847 CrossRef CAS PubMed.
- Y. Kawakami, Y. Sakuma, T. Wakuda, T. Nakai, M. Shirasaka and Y. Kabe, Organometallics, 2010, 29, 3281 CrossRef CAS.
- K. Kawahara, H. Tachibana, Y. Hagiwara and K. Kuroda, New J. Chem., 2012, 36, 1210 RSC.
- I. Hasegawa, K. Ino and H. Ohnishi, Appl. Organomet. Chem., 2003, 17, 287 CrossRef CAS PubMed.
- N. Xu, E. J. Stark, P. R. Dvornic, D. J. Meier, J. Hu and C. Hartmann-Thompson, Macromolecules, 2012, 45, 4730 CrossRef CAS.
- J. Beckmann, D. Dakternieks, A. Duthie, M. L. Larchin and E. R. T. Tiekink, Appl. Organomet. Chem., 2003, 17, 52 CrossRef CAS PubMed.
- Y. Hagiwara, A. Shimojima and K. Kuroda, Chem. Mater., 2008, 20, 1147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Fig. S1–S12. See DOI: 10.1039/c5cc03668b |
| This journal is © The Royal Society of Chemistry 2015 |