
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlue-emitting pyrene-based aggregates†
Jorge S.
Valera‡
a,
Joaquín
Calbo‡
b,
Rafael
Gómez
a,
Enrique
Ortí
*b and
Luis
Sánchez
*a
aDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: lusamar@quim.ucm.es
bInstituto de Ciencia Molecular, Universidad de Valencia, 46980 Paterna, Spain. E-mail: enrique.orti@uv.es
First published on 13th May 2015
Abstract
The supramolecular polymerization of pyrene imidazoles 1 and 2, governed by H-bonding and C–H⋯π interactions, yields aggregates showing the characteristic bluish emission pattern of pyrene-based monomers.
Since its discovery in 1837 by Laurent,1 pyrene has become one of the most studied polycyclic aromatic hydrocarbons due to its attractive electronic and photophysical properties, and it has been widely tested as an active component in organic electronics.2,3 Pyrene is a blue-emitting material with a tendency to form excimers both in solution and in the solid state. The formation of excimers red-shifts the emission and reduces its intensity.2–4 To improve the emissive properties, great efforts have been devoted to the structural modification of pyrene to avoid aggregation and to suppress the quenching effect of the π–π interactions. Therefore, a challenging task in pyrene chemistry is the design of derivatives in which supramolecular order and appropriate blue-emitting features can coexist.
Herein, we describe the synthesis and the supramolecular polymerization of the pyrene imidazoles 1 and 2 endowed with peripheral achiral and chiral alkyl chains, respectively (Fig. 1).41 and 2 have been designed with a variety of supramolecular interaction motifs that guide their self-assembly through H-bonding interactions between amide groups and imidazole moieties together with a number of C–H⋯π dispersion interactions. The organized supramolecular structures are able to immobilize apolar solvents like cyclohexane forming gels.5 Notably, the aggregates formed by 1 and 2 show the characteristic blue-emitting pattern of pyrene-based monomers.
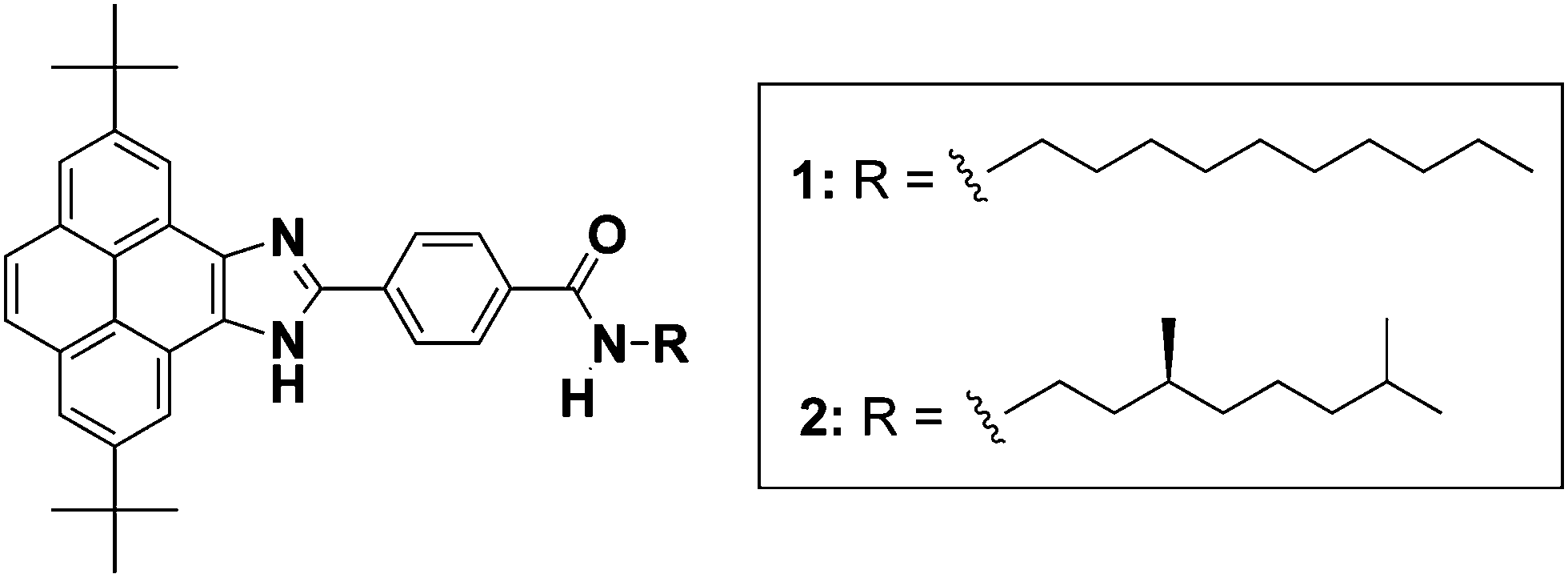 | ||
| Fig. 1 Chemical structure of pyrene imidazoles 1 and 2. | ||
The preparation of 1 and 2 was accomplished starting from pyrene, 4-carboxybenzaldehyde and the corresponding amine (Scheme S1, ESI†).6 Prior to the synthesis of the final derivatives, two tert-butyl groups were incorporated at positions 2 and 7 of the pyrene moiety to increase the solubility of the target compounds. The chemical structure of 1 and 2 was confirmed by NMR, FT-IR and HRMS analysis (see the ESI†).
To unravel the supramolecular polymerization mechanism of 1 and 2, temperature-dependent UV-Vis spectroscopy measurements were performed in methylcyclohexane (MCH) as solvent (Fig. S1 and S2, ESI†). The bands observed at around 355 and 400 nm in the UV-Vis spectra of both 1 and 2 experience a slight bathochromic shift and a broadening upon decreasing the temperature, which is diagnostic of the aggregation process. Plotting the variation of the absorbance at 404 nm with the temperature results in cooling curves with an apparent sigmoidal shape, characteristic of an isodesmic supramolecular polymerization mechanism (Fig. S1b and S2b, ESI†).7
To obtain a complete set of thermodynamic parameters, the UV-Vis cooling curves derived for 1 and 2 were fitted to the model recently reported by ten Eikelder et al.8 The thermodynamic parameters obtained for 1 and 2 demonstrate that the branched nature of the peripheral alkyl chain in 2 plays a relevant role, showing significantly different values of the enthalpy of elongation (ΔHe), the entropy of elongation (ΔS) and the nucleation penalty (ΔHn) (Table 1). In contrast, the total free energy (ΔG) is very similar in both pyrene derivatives (∼35 kJ mol−1). Especially interesting is the calculated value of the degree of cooperativity σ. Despite the sigmoidal shape of the cooling curves, σ presents values of 5.5 × 10−2 and 9.3 × 10−3 for compounds 1 and 2, respectively, which denote a certain degree of cooperativity in the supramolecular polymerization of these pyrene derivatives (Table 1). Interestingly, the self-assembling properties of compounds 1 and 2 are similar and both readily form gels in cyclohexane at a concentration of ca. 1 wt%. The organogels exhibit blue emission upon irradiation at 364 nm (Fig. 2).
| 1 | 2 | |
|---|---|---|
| a The nucleation penalty ΔHn is negative, which implies that the enthalpy gain is smaller for nucleation compared to elongation. b The equilibrium constants for elongation (Ke) and dimerization (Kn), and the cooperativity factor (σ = Kn/Ke) are calculated at 298 K. | ||
| ΔHe [kJ mol−1] | −72 ± 1 | −52 ± 1 |
| ΔS [J Kmol−1] | −122 ± 3 | −58 ± 2 |
| ΔHn [kJ mol−1]a | −7.1 ± 0.2 | −11.5 ± 0.2 |
| ΔG [kJ mol−1]a | −35.88 | −34.73 |
| σ [—]b | 5.5 × 10−2 | 9.3 × 10−3 |
| K e [L mol−1]b | 2.04 × 106 | 1.5 × 106 |
| K n [L mol−1]b | 1.13 × 105 | 1.21 × 104 |
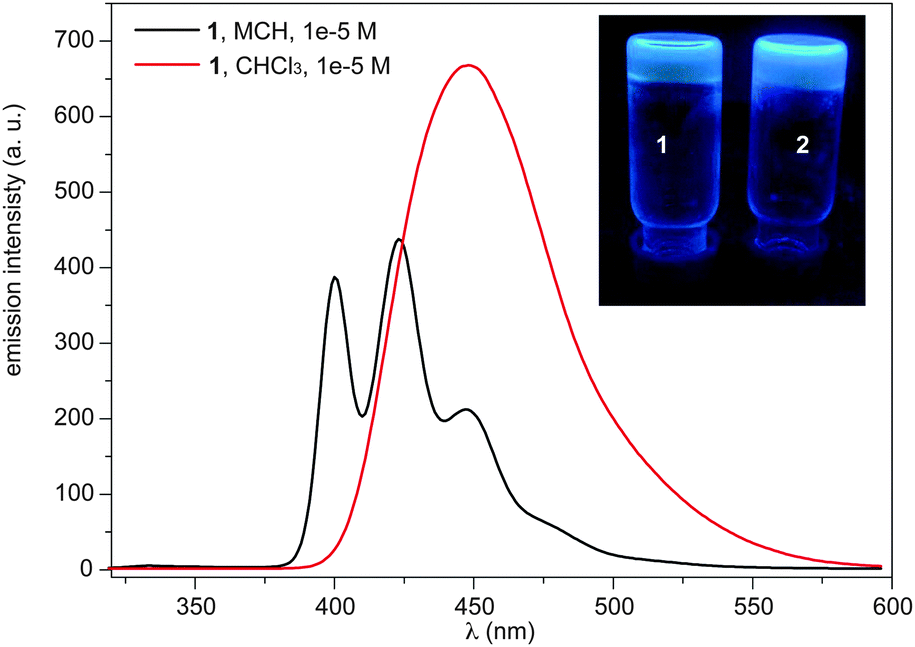 | ||
| Fig. 2 Emission spectra of 1 in MCH and CHCl3 (298 K, 1 × 10−5 M, λexc = 334 nm). The inset shows a picture of the cyclohexane gels obtained from compounds 1 and 2 with illumination at 364 nm. | ||
The emission properties of 1 and 2 were investigated in CHCl3 and MCH. The emission spectra in MCH at 1 × 10−5 M, conditions under which 1 and 2 are aggregated, show the typical pattern of monomeric pyrene with maxima at 400, 423 and 447 nm (Fig. 2 and Fig. S3, ESI†). In contrast, the emission spectra in CHCl3, a good solvent that facilitates the solvation of isolated molecules, at 1 × 10−5 M feature broad and structureless bands centred at 448 nm, evidencing the formation of pyrene excimers (Fig. 2 and Fig. S3, ESI†).9,10 Compounds 1 and 2 show a remarkable fluorescence quantum yield (ϕf) in the aggregated state (MCH, 1 × 10−5 M) of 0.43. A lower ϕf value is measured for the excimer formed in CHCl3 (0.38 and 0.22 for 1 and 2, respectively). To the best of our knowledge, this unusual emissive behaviour has only been described for a pyrene derivative endowed with a chiral oligo(glutamic acid) side chain,11 and it was ascribed to a helical supramolecular organization.
The presence of the stereogenic center on 2 was utilized to put in evidence the possible helical organization of the aggregates formed by 1 and 2 in an attempt to justify the observed emission properties. Compound 2 shows a weak dichroic response with a poorly defined bisignated Cotton effect centered at around 250 nm (Fig. S5a, ESI†). In addition, sergeants-and-soldiers (SaS) experiments were performed by mixing achiral 1 and chiral 2 to investigate the amplification of chirality.12 The SaS experiment shows a linear increase of the dichroic response upon increasing the amount of chiral sergeant 2 (Fig. S5b, ESI†). The dichroic data reveal that compounds 1 and 2 do not self-assemble forming helical structures and, therefore, this supramolecular organization cannot be invoked to justify the observed emission features.
The experimental data arise two questions: (1) which are the non-covalent forces participating in the self-assembly of 1 and 2? and (2) what kind of supramolecular aggregate is formed upon their self-assembly capable of exhibiting the above-mentioned emissive features? To give an appropriate answer to these questions, experimental and theoretical studies were performed. In the FTIR spectra of 1 and 2, the amide I and amide II bands at around 1635 and 1560 cm−1, diagnostic of the formation of H-bonds between the amides, are visible (Fig. S6, ESI†).13 In the concentration-dependent 1H NMR experiments, the triplet at δ ∼ 6.3, corresponding to the amide groups, and the resonance corresponding to the imidazole N–H group (δ ∼ 13) show a downfield shift with increasing concentrations that is indicative of the formation of an array of H-bonds (Fig. S7, ESI†). On the other hand, the aromatic resonances, used to envisage the intermolecular interactions between the aromatic fragments, show an opposite shifting of the δ values upon increasing the concentration. Thus, whilst the resonance of pyrene at δ ∼ 9 experiences a shielding effect, one of the anisochronous protons at the para position of the benzamide moiety (δ ∼ 8.2) shifts to lower δ values. A rotating-frame Overhauser effect spectroscopy (ROESY) NMR experiment in a concentrated solution of 1 (45 mM, CDCl3, 298 K) was therefore conducted to extract more information about the supramolecular organization of this pyrene derivative.14
The ROESY experiment shows a number of intramolecular through-space coupling signals between the protons spatially close within the molecule (dotted blue circles in Fig. 3). In addition, ROESY signals corresponding to intermolecular contacts are also observed (dotted green lines in Fig. 3). Especially interesting are the contacts observed between the methylene units of the alkyl side chain and most of the aromatic protons of the pyrene unit. These contacts suggest an alternated distribution of the pyrene units in the aggregated state that could justify the observed monomeric emission properties.
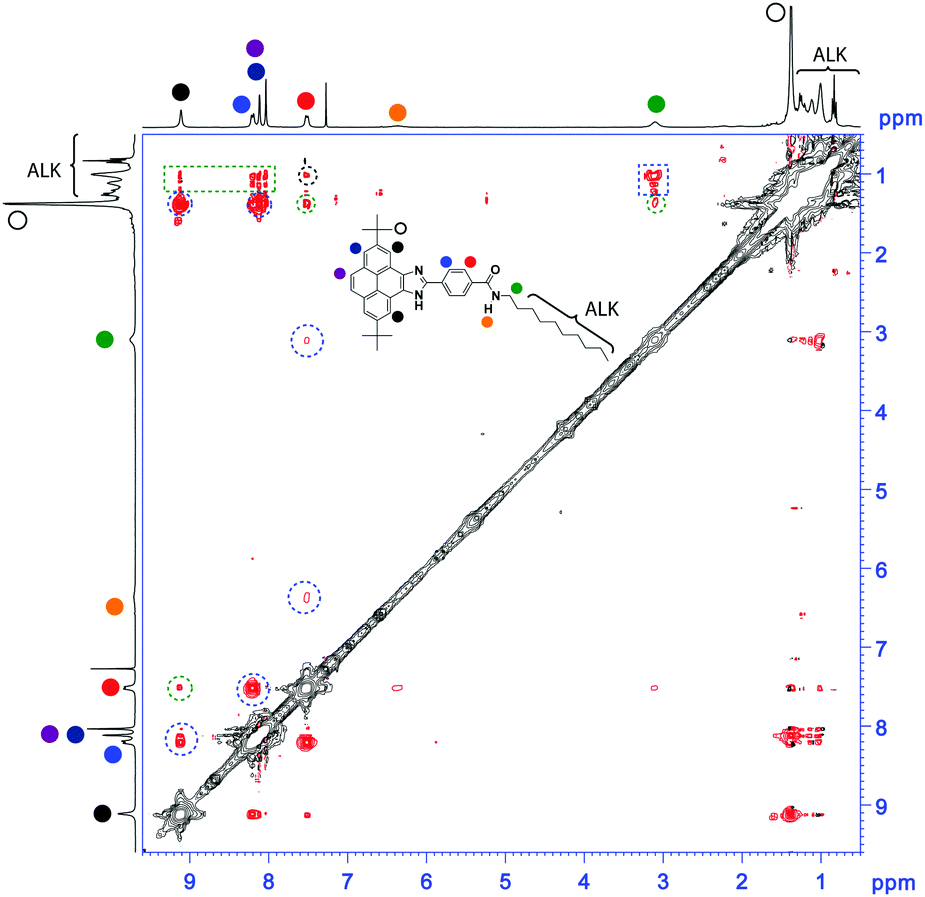 | ||
| Fig. 3 ROESY NMR spectrum (CDCl3, 300 MHz, 45 × 10−3 M, 293 K) of 1. The dotted blue, green and black lines highlight intra-, intermolecular or both through-space coupling signals, respectively. The inset shows the chemical structure of 1 depicting the anisochronous protons by coloured circles. | ||
Theoretical calculations were performed on compound 1 within the density functional theory (DFT) framework to shed light on the structural and energetic aspects of the pyrene imidazole system upon aggregation (see the ESI† for computational details). The isolated molecule shows a planar minimum-energy structure with an out-of-plane disposition of the amide group (Fig. S8, ESI†) that facilitates H-bonding interactions between neighbouring molecules. To unravel the possible contribution of the different parts of the molecule in the operation of non-covalent forces, the molecular electrostatic potential (MEP) calculated for 1 (Fig. S9, ESI†) was analysed. The MEP surface shows that the largest potential energy points are concentrated in the amide and the imidazole groups. Negative potential energies of −55.7 and −54.6 kcal mol−1 are calculated in the oxygen and nitrogen vicinity, respectively, whereas positive potentials of +101.2 and +121.0 kcal mol−1 are located in the N–H region of the amide and imidazole, respectively. Strong electrostatic interactions are then expected between monomers by means of H-bonding interactions involving the amide and imidazole units, as well as weak π–π and/or C–H⋯π dispersion interactions between the aromatic units and the aliphatic chains.
A number of dimers, as minimal supramolecular units, can be modelled for 1 owing to the variety of supramolecular interaction motifs present in the monomer: (i) the aromatic structure, (ii) the polar amide and imidazole groups and (iii) the nonpolar long aliphatic chain. In a first approach, two monomers may accommodate either in a parallel or antiparallel disposition by the π-stacking of the pyrene imidazole cores with themselves or with the benzamide group, respectively (dimers A and B in Fig. 4). In dimer A, an amide–amide H bond is formed due to the rotation of ∼30° of the upper monomer around the perpendicular axis. This structure would grow up in a helical column, but this possibility has been discarded by CD measurements. Similar to A, dimer C is arranged in a parallel mode but with the amide groups pointing in opposite directions and forming a non-linear H-bond (Fig. 4). A weak H-bonding N–H⋯N interaction between the imidazole rings is also present in antiparallel dimer B.
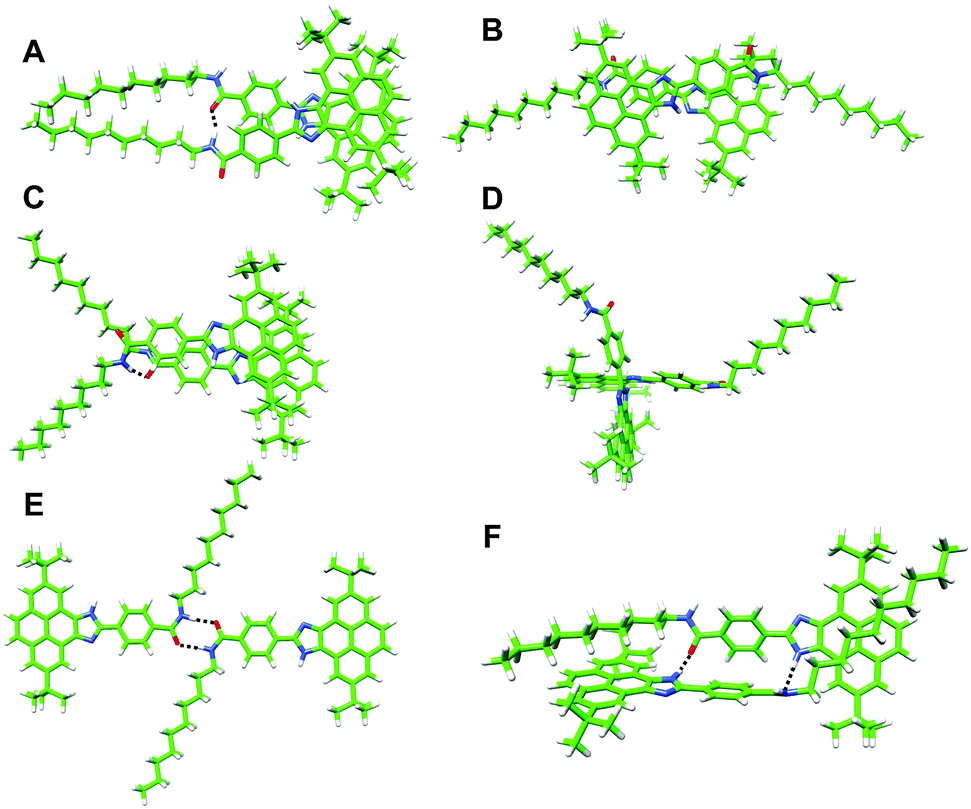 | ||
| Fig. 4 Minimum-energy geometries calculated for the different dimers postulated for 1 at the B97D3/6-31G** level of theory. | ||
Besides the π–π recognition motif that gives rise to structures A–C, the polar imidazole and amide groups constitute high potential energy regions for the supramolecular stabilization of the dimers. Thereby, dimers D–F, in which the formation of strong H-bonds is the recognition driving force, have been modelled (Fig. 4). Whereas dimers D and E are only stabilized by the H-bond interaction between the imidazole and the amide groups, respectively, dimer F forms two H-bonds between the amide and the imidazole groups together with an extended CH⋯π stabilization due to the interaction between the aliphatic chains and the pyrene imidazole cores.
Table 2 summarizes the interaction energy (Eint) and the main intermolecular forces governing the self-assembly of dimers A–F calculated at the B97D3/6-31G** level. A topological analysis of the electron density was also carried out by means of the NCIPLOT software to obtain the non-covalent interaction (NCI) surfaces, which provide additional information on the origin of the supramolecular aggregation (Fig. S10 and S11, ESI†).
| E int (kcal mol−1) | n HB | d HB (Å) | π–π | CH⋯π | CH⋯CH | |
|---|---|---|---|---|---|---|
| A | −54.6 | 1 | 1.918 | ✓ | — | ✓ |
| B | −51.0 | 1 | 2.064 | ✓ | — | — |
| C | −53.7 | 1 | 2.051 | ✓ | — | — |
| D | −32.0 | 1 | 1.826 | — | ✓ | — |
| E | −7.7 | 2 | 1.822 | — | — | — |
| F | −61.7 | 2 | 1.839 | — | ✓ | — |
Dimers A, B and C are computed very close in energy owing to the similar number and nature of the interactions stabilizing the aggregate. Dimers B and C display the typical green surface indicative of dispersion interactions along the π–π skeleton between monomers (Fig. S10, ESI†). Stabilizing CH⋯CH interactions between the aliphatic chains are additionally found for A, which is calculated to be slightly more stable than B and C. In these three dimers, weak H-bonds are formed as indicated by the closest NH⋯N/O contacts in the range of 1.92–2.06 Å (Table 2).
The situation is different for dimers D, E and F. Dimer E is stabilized by two H-bonding interactions between the two amides in a cis disposition. The occurrence of dimer E is however very unlikely because of: (i) the high barrier (15 kcal mol−1) that has to be overcome to convert a trans amide into a cis amide (Fig. S12, ESI†) and (ii) the small interaction energy (−7.7 kcal mol−1) resulting from the lower stability of the cis conformation. The cross shape of dimer D enables a strong H-bond interaction between the imidazole groups (N⋯H–N distance of 1.83 Å) and incorporates C–H⋯π interactions between the tert-butyl groups of one monomer and the aromatic pyrene core of the other (Fig. S10, ESI†). Nonetheless, the Eint computed for this dimer (−32.0 kcal mol−1) cannot compete with the stabilization obtained for the π–π patterned dimers A–C. Finally, dimer F combines H-bonding interactions between the imidazole and amide groups and C–H⋯π stabilizing interactions between the alkyl chains and the pyrene cores (Fig. 4 and Fig. S10, ESI†). The additive effect of these interactions leads to the highest stability (Eint = −61.7 kcal mol−1) among all the computed dimers. The structure of dimer F allows us to identify all the ROESY signals observed experimentally and, in particular, the short contacts between the alkyl chains and the pyrene cores (Fig. S13, ESI†). In addition, the absence of π–π interactions between the pyrene cores in dimer F explains the emission properties similar to the monomer. Dimer F is therefore proposed as the most plausible associate for compounds 1 and 2.
To rationalize how dimer F can grow up from such nonplanar, distorted disposition of the monomers, the tetramer shown in Fig. 5 was built up through an imidazole–amide–imidazole H-bond sequence. The minimum-energy geometry computed for the tetramer reveals that the H-bonding motifs are preserved along the monomeric pairs. The average NH(amide)–N(imidazole) contact is computed to be 2.18 Å, whereas the CO(amide)–HN(imidazole) interaction is calculated to be 1.82 Å. The total interaction energy in the tetramer reaches −182.1 kcal mol−1, which corresponds with an Eint = −60.7 kcal mol−1 per monomer pair. This value is very close to the Eint computed for dimer F (−61.7 kcal mol−1), suggesting that: (i) the oligomer can easily grow keeping the strong H-bond pattern and the stabilizing C–H⋯π forces and (ii) the degree of cooperativity in the supramolecular polymerization should be very small.
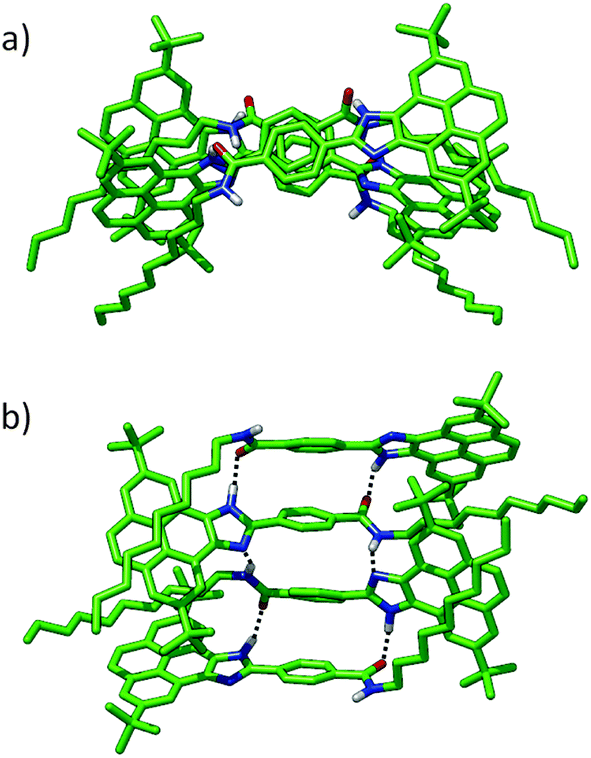 | ||
| Fig. 5 Top (a) and side (b) views of the B97D3/6-31G**-optimized geometry calculated for the tetramer of 1. Dotted lines emphasise the H-bonding array established between the imidazole and amide units. Only hydrogen atoms involved in H-bonding interactions are shown. | ||
In summary, we have synthesised pyrene imidazoles 1 and 2 that prevent the formation of excimers upon aggregation. The number and type of non-covalent interactions operating in the aggregation of these pyrene derivatives result in an amazing scenario of possibilities for 1 and 2 to self-assemble. Both experimental and theoretical studies demonstrate that the formation of the aggregates is mainly governed by the H-bonding interaction between the amide functionality and the imidazole moiety, together with a number of C–H⋯π interactions. The absence of π–π interactions between the pyrene cores determines that the aggregate preserves the blue-emitting properties of the monomer. The results presented herein demonstrate the complex pathway followed by 1 and 2 to finally yield blue-emitting supramolecular aggregates.
Financial support from the MINECO of Spain (CTQ2011-22581 and CTQ2012-31914), European Feder funds (CTQ2012-31914), UCM (UCM-SCH-PR34/07-15826), Generalitat Valenciana (PROMETEO/2012/053) and Comunidad de Madrid (NanoBIOSOMA, S2013/MIT-2807) is acknowledged. JC thanks the MECD for a doctoral FPU grant.
Notes and references
- A. Laurent, Ann. Chim. Phys., 1837, 66, 136 Search PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed.
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260 CrossRef CAS PubMed.
- (a) N. Tchebotareva, X. M. Yin, M. D. Watson, P. Samori, J. P. Rabe and K. Müllen, J. Am. Chem. Soc., 2003, 125, 9734 CrossRef CAS PubMed; (b) D. Venkataraman, S. Lee, J. S. Zhang and J. S. Moore, Nature, 1994, 371, 591 CrossRef CAS PubMed.
- (a) A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109 RSC; (b) J. W. Steed, Chem. Soc. Rev., 2010, 46, 3686 RSC.
- D. Kumar, K. R. J. Thomas, C.-C. Lin and J.-H. Jou, Chem. – Asian J., 2013, 8, 2111 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed.
- H. M. M. ten Eikelder, A. J. Markvoort, T. F. A. de Greef and P. A. J. Hilbers, J. Phys. Chem. B, 2012, 116, 5291 CrossRef CAS PubMed.
- F. M. Winnik, Chem. Rev., 1993, 93, 587 CrossRef CAS.
- The emission corresponding to the pyrene monomer is only reached in CHCl3 by using highly diluted (7 × 10−9 M), deoxygenated solutions (Fig. S4, ESI†).
- Y. Kamikawa and T. Kato, Langmuir, 2007, 23, 274 CrossRef CAS PubMed.
- (a) M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606 CrossRef CAS PubMed; (b) F. García, P. A. Korevaar, A. Verlee, E. W. Meijer, A. R. A. Palmans and L. Sánchez, Chem. Commun., 2013, 49, 8674 RSC.
- (a) Y. Matsunaga, N. Miyajima, Y. Nakayasu, S. Sakai and M. Yonenaga, Bull. Chem. Soc. Jpn., 1988, 61, 207 CrossRef CAS; (b) F. García, P. M. Viruela, E. Matesanz, E. Ortí and L. Sánchez, Chem. – Eur. J., 2011, 17, 7755 CrossRef PubMed.
- Prior to this ROESY experiment, the aggregation of 1 was assessed by diffusion-ordered spectroscopy (DOSY) NMR, which reveals a larger value of the diffusion coefficient when compared to a more diluted solution (Fig. S6b, ESI†). A. Wong, R. Ida, L. Spindler and G. Wu, J. Am. Chem. Soc., 2005, 127, 6990 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S12, theoretical computational details and experimental section. See DOI: 10.1039/c5cc03616j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |